This blog is written after consulting many interviewers and these are the common questions which are being asked in clinical research job related interviews. You can understand after reading this, that almost all questions are related to basics of clinical research. It is not possible to provide in depth details of each topic so we provide the links to each questions so that you can read in details.
Table of Contents
What is informed consent?
Before determining whether to participate, informed consent is the method of discovering crucial information about a clinical trial. The provision of information for the participants is also a continuous process during the study.
The doctors and nurses involved in the trial explain the details of the study to help someone decide whether to participate or not. If the participant’s native language is not English, translation assistance can also be provided.
The research team also offers an informed consent document that contains specifics of the project, such as its purpose, duration, required procedures, and key contacts.
More at: https://lifepronow.com/2020/07/03/informed-consent-defination-and-informed-consent-form-example/
The informed consent statement outlines risks and potential benefits. Then the person decides whether to sign the contract or not. Informed consent is not a contract, and the defendant will withdraw at any time from the trial.
What is Primary and Secondary End points
Primary end point are the specific event that the study is designed to assess the effect of the drugs upon. It answers the most important question of the clinical trial. They are also called primary outcome measure.
Secondary end point are additional events of interest, but which the study is not specifically powered to assess. They are also knows as Secondary outcome measures.
More at: https://lifepronow.com/2020/06/29/primary-endpoint-and-secondary-endpoint/
Orphan Drug Designation in US, EU and Japan
The Drug approval process in EU
Drug Approval Process in India
What is treatment arm?
A group of subjects that receives a specific intervention/treatment, or no intervention in clinical trial. Group of subjects who receive treatment, are called treatment arm.
Active comparator arm:Group of subjects receive standard treatment
Placebo comparator arm: Group of subjects receive standard treatment
Sham comparator arm: group of subjects receive same procedure or device without active component or process. (It is like Placebo comparator arm but for medical devices and procedure)
What is Surrogate end point?
As per National Cancer Institute: “In clinical trials, an indicator or sign used in place of another to tell if a treatment works. Surrogate endpoints include a shrinking tumor or lower biomarker levels.”
Alternatively of stronger metrics such as longer lifespan or better quality of life, they could be used as the test results can be assessed earlier.
Using surrogate endpoints in clinical trials that allow for earlier approval of new drugs for treating severe or life-threatening diseases , such as cancer. Surrogate endpoints are not necessarily accurate markers or measures of how well a treatment operates.
More at: https://lifepronow.com/2020/07/05/surrogate-endpoint-marker-definition-and-examples/
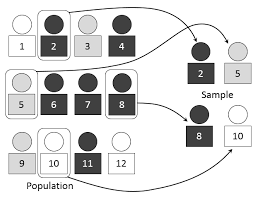
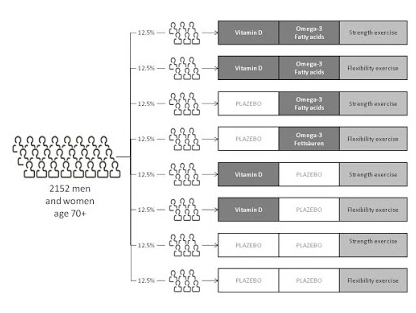
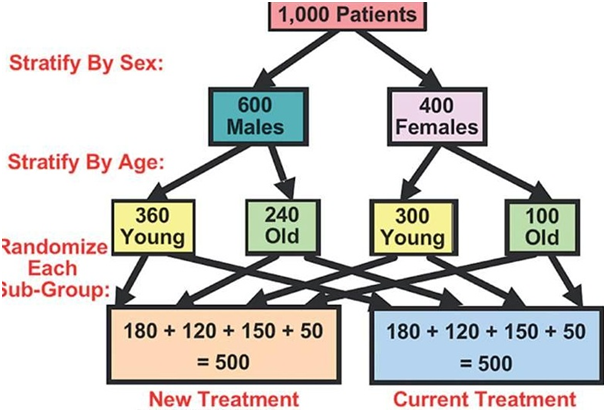
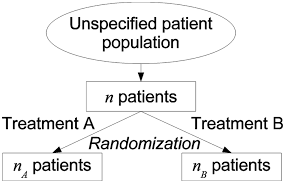
What do you understand by Randomization?
As per the National Cancer Institute “In research, the process by which participants in clinical trials are assigned by chance to separate groups that are given different treatments or other interventions.
Neither the researcher nor the patient chooses what medication or intervention they should receive.
Using chance of assigning people to groups means that the results of the care or intervention obtained by the participants can be more equally compared.
More at: https://lifepronow.com/2020/07/13/randomization-in-clinical-trialsdefinition-and-types/
What do you know about ICH and GCP?
The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use is the full form of ICH.
Guidelines provided by ICH can be divided into 4 categories: Quality (Q), Safety (S), Efficacy (E) and multidisciplinary guidelines (M).
These guidelines help to harmonize technical guidelines which are being followed by many countries for drug registration.
Read about missing and member countries: https://www.ich.org/
GCP’s full form is good clinical practice. It is provided by ICH.
ICH E6 (R2) segment provides detailed information about GCP. Good clinical practice offers a system of guidelines to ensure the health of participants in the study and the quality and validity of data. GCP contains 13 principles which can be studied here. https://ichgcp.net/2-the-principles-of-ich-gcp-2
Where do the ideas for trials come from?
Ideas typically come from researchers for the clinical trials. After the researchers test new therapies or procedures in the laboratory and in animal studies, the experimental treatments with the most promising laboratory results are moved into clinical trials.
More and more information are gained during a trial about an experimental treatment, its risks and how well it may or may not work. But In general practice, It is decided as organization level.
Organization decides the therapeutic areas in which it wants to develop the molecule. And this is decided based on various factors such as market size, competition, target country, nature of disease etc.
What are the different types of clinical studies?
There are two main type of trials: Observational clinical trial and interventional clinical trial
“Interventional clinical study is performed with the purpose of studying or representing clinical or pharmacological properties of drugs/devices, their side effects and to establish their efficacy or safety.”
Whereas observational studies,are those studies where intervention is not given (treatment, drug, surgery). There are many type of clinical studies and you can read about each study in detail here: (case report or case series, ecologic, cross-sectional (prevalence study), case-control and cohort studies)
More at: https://lifepronow.com/2020/05/12/clinical-trial-introduction-types/
Interviewer can ask about each individual study type as well. so read all the sub-types as well in details.
What are the Phases of clinical trials?
Clinical trials are conducted in four phases. Each Trial have different purpose and help scientists answer different questions:
In Phase I trials, researchers test an experimental drug or treatment in very small group of population (20-80) for the first time in order to evaluate its safety, determine a safe dosage range, and to identify side effects. (Maximum tolerated dose assessment)
In Phase II trials, the experimental study drug or treatment is given to a larger group of population (100-300) to see if it is effective and to further evaluate its safety. (Efficacy)
In Phase III trials, the experimental study drug or treatment is given to large groups of population (1,000-3,000) to validate its effectiveness, monitor side effects, compare it to commonly used treatments, and assemble information that will allow the experimental drug or treatment to be used very safely. (effectiveness)
In Phase IV trials, post-marketing studies describe additional information including the drug’s risks, benefits, and best possible use. (Post Marketing surveillance)
More at: https://lifepronow.com/2020/05/11/phases-of-clinical-trials/
What is “expanded access”?
Most human use of the investigational new drugs takes place in controlled clinical trials conducted to measure the safety and efficacy of the new drugs.
Data from these trials are used in order to determine whether a drug is safe and effective, and provide as the basis for the drug marketing application.
Occasionally, patients do not qualify for these controlled trials because of other health problems, age, or other factors, or are or else unable to enroll in such trials (e.g., a patient may not live sufficiently close to the clinical trial site).
For patients who are unable to enroll in a clinical trial of an investigational drug but have a significant disease or disorder that could benefit from the drug therapy, FDA regulations require suppliers of these medications to provide certain patients with access to the medication in such cases, known as “expanded access.
“For example, the drug can’t expose patients to unreasonable risks given the severity of the disease to be treated and the patient does not have any other satisfactory therapeutic options (e.g., an approved the drug that could be used to treat the patient’s disease or condition).
The manufacturer must be willing to make the drug available for the expanded access use. The primary intention of expanded access is to provide treatment for the patient’s disease or condition, rather than to collect data about the study drug.
Many investigational medications are licensed from prescription suppliers for patient use through extended access services listed in ClinicalTrials.gov. If you or a loved one are interested in treating an investigational drug under an expanded access protocol listed in ClinicalTrials.gov, review the eligibility criteria for the protocol and ask at the Contact Information Number.
What do you know about single and double blind studies?
In a single blind study, the subjects in the clinical trial do not know if they are receiving the placebo or the real treatment. where as in double blind clinical trial , both the subjects and the investigator do not know which group got the placebo and which got the experimental treatment.
What are Triple Blind Studies?
In triple blind studies, Subjects, Investigator and individuals who are responsible for analysis the outcomes are blinded. You can find the example of triple blind study here:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4400707/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1118396/
You should also read about the benefits and disadvantages of blinding.
What is Open label trials?
Open label trials are those trials where no blinding is done.
Subjects and Principle investigator and outcome analysis team knows about treatment and placebo group.
What is Placebo?
“A placebo is anything that seems to be a “real” medical treatment — but it doesn’t. It might be a pill, a shot, or some other type of “fake” treatment. All placebos have one thing in common is that they do not contain an active substance meant to affect health.
Normally, the person getting a placebo doesn’t know for sure that the treatment isn’t real.
The placebo is often in the form of a “sugar pill,” but it may also be an injection, a liquid, or even a procedure. This is intended to act like a true cure, but it does not affect the disease directly.
https://lifepronow.com/2020/07/08/placebo-is/
What is the difference between IB and Protocol
Clinical trial protocol describes the objectives, design, methodology, statistical considerations and aspects related to the organization of clinical trials.”
In simple words, We can say clinical trial protocol tells us “how to conduct scientifically sound, reliable and ethically compliance trials”
“Investigator Brochure enables investigators (Doctor at hospital) conducting clinical studies to know the risks and benefits associated with an investigational product.”
What is the approval process by FDA
The regulatory agency of USA is FDA which has two bodies: CBER and CDER.
CBER stands for Centre for biological evolution and Research and CDER stands for Centre for drug evaluation and research. CBER agency deals with biological product whereas CDER deals with drugs and biological both. In general the drug (chemical) approval process in US can be divided into four parts, they are:
- Supplemental new drug application
- Abbreviated new drug application
- Petitioned ANDA
- and New drug application
Details about each process, you can read here:
https://lifepronow.com/2019/12/21/drug-approval-process-nda-snda-anda-fda/ - If you are preparing for regulatory affair interview, then you can further read this topic in details here:
Drug Approval Process in India
Approval pathways of Fixed dose combination (FDC) in India
Orphan Drug Designation in US, EU and Japan
Biologics Approval Pathways in the USA:
Medical Device Registration in Japan-Todokede, Ninsho and Shonin Pathways
Generic Products Approval Pathways (USA): Paragraph Certification I, II, III, and IV:
Drug Approval Process in China
What is SAP
SAP (Statistical Analysis Plan) is the document that contains detailed information about the statistical methods and the study objectives to help in the production of the Clinical Study Report (CSR) that includes figures, summary tables, and subject data listings for Protocol.
Documentation of the program variables and algorithms which would be used to produce descriptive statistics and statistical analysis.
What is LOCF?
Pharmaceutical companies spend numerous months to conduct longitudinal studies on the human subject. It is impractical to expect patients to keep timely visit over such a long period of time.
In spite of all the efforts, patient data are not collected for some and these become missing values in a SAS data set soon.
The most recent previously available value for each of the missed visits is replaced for reporting. This is classified as Carried Forward Last Observation (LOCF).
LOCF does not mean the last SAS dataset observation which gets carried forward rather it means last non-missing value carried forward. It is the values of the individual measures which are actually “observations” in this case.
Also if there are multiple variables containing these values then they will be carried forward separately.
Explain the role of clinical research coordinator
In general, clinical research coordinator works as an assistant to Principle investigator (PI). CRC helps in taking the informed consent forms, enter the the in the eDC, followup with the subjects for site visits, respond to data management and clinical research associate queries. They are also involved in budget planning and management of budgets for the study and financial payments.
Details: https://lifepronow.com/2020/05/16/beginners-guide-to-clinical-research-coordinator/
You can also explore the role of CRA here, which is similar question:
What is the difference between Effectiveness and Efficacy
“Efficacy = best possible results for a particular intervention, under perfect conditions;
“Effectiveness = most likely results for a particular intervention under pragmatic or ‘real-life’ conditions (taking into account compliance, dropouts, withdrawals, the learning curve for surgical interventions, etc.)
more at https://lifepronow.com/2020/05/18/efficacy-vs-effectiveness/
What Are The Benefits And Risks Of Participating In Clinical Trial?
Benefits include: Playing an active role in one’s health care, having access to medications that may not be accessible for a significant period of time, and supporting people by engaging in the study so that the drug can eventually be accepted and made readily available.
Risks include: Participation in the clinical trial may involve some risks that doctor will explain in more detail. These risks may include:
• Side-effects that are known and those that have not yet been identified;
• risks associated with the study procedures;
• the experimental treatment may be not be effective or less effective than the current standard;
• the experimental treatment may not work for all patient.
Additionally, in some clinical trials the patient may not receive the experimental treatment, although the current standard or a placebo.
In addition to the risks listed above, the trial might require all the participant’s time and attention—including trips to the study site, more treatments, hospital stays or complex dosage requirements.
A prospective, randomized, double-blind, controlled clinical trial is the most rigorous clinical trial design, and the one that regulatory agencies mandate must be conducted to demonstrate a medication’s effectiveness and safety.
Those trials are the highest quality evidence on the medication and its activities in a new drug application, which form the basis for approval.
Patients are carefully chosen for inclusion in this research design and are randomly allocated to obtain the experimental treatment or a similar active drug or placebo.
Neither the patient nor the treating physician knows what care was offered, thereby eliminating potential bias.
Individual definitions of the study descriptions are:
Prospective: Looking ahead, starting before therapy has started.
Randomized: Patients are randomly assigned to obtain the alternative or experimental treatment ( e.g., standard of care or placebo)
Double-blind: Neither patients nor research personnel know which participants receive the experimental drug and which participants receive a placebo or normal treatment.
Controlled: One group of the patients will be given an experimental drug or treatment, while a second group is given either the standard treatment for the illness or a placebo.
Who Sponsors A Clinical Trial?
Clinical trials can be sponsored or funded by a variety of the organizations or individuals including physicians, medical institutions, voluntary groups, foundations, and pharmaceutical companies, in addition to the government agencies such as the National Institutes of Health (NIH), the Department of Defence (DOD), Human Health and Services (HHS), and Department of Veteran’s Affairs (VA).
What should people consider before participating in a trial?
People will learn about the clinical trial as best as possible and feel comfortable asking health care team members questions about it, the treatment provided when in a trial, and the cost of the experiment.
The following questions may be of assistance to the participant to discuss with the health care team. Some of the answers to these questions are found in the informed consent document.
- What is the purpose of study?
- Who is going to be in study?
- How do researchers think the experimental treatment being tested can be effective? Is it reviewed in advance?
- Which kinds of tests and experimental treatments?
- What does the research equate the potential complications, side effects and benefits with my current treatment?
- Why does the case affect my daily life?
- How long is the case going to last?
- Does it take hospitalisation?
- What will pay for the experimental treatment?
- Would I get a refund for other expenses?
- Which sort of long-term follow-up treatment does this study include?
- Why do I know the experimental medication is functioning?
- Will it provide me with the results of the trials?
- Who will take the responsibility for my care?
What is the scope of clinical research in India?
The Scope of Clinical Research depends on the growth of the Medical / Pharmaceutical Industry. And in India, this growth is immense, and the pace is rising with time, due to:
– an ageing population
– new developments in treatments and diagnosis
– the Indian economy – its privatization, liberalization & globalization.
Drug discovery involves designing new drugs, and after identifying a prospective article, the drug development process takes place. Action, safety, use, formulation, quality control, storage, packaging, and marketing are few of the other stages associated with the drug development process.
The global demand is more than $70 trillion for this knowledge-intensive field. And it is projected that India ‘s contribution is about 15 percent of the global market, which is about $10.5 billion. The business is currently rising at an average annual cumulative growth rate of a
And it is this reach that attracted Pharmaceutical Companies & Contract Research Organizations (CROs) to India to develop their research offices or even forward some of their clinical activities, such as data processing or quality assurance, to other CROs working in India, thereby reducing costs.
Clinical Research is an field of highly exclusive & lucrative job prospects with the potential to increase your salary in less than a decade by 400 per cent.
In order to soar in the evergreen industry, one should train oneself through appropriate courses that allow candidates to be ready for various operations of clinical research.
When you are interested, you will consider these internship-led training programmes that are conducted at MMS University (www.mmsuniversity.com), MMS Holdings’ educational division (www.mmsholdings.com), in fields these as Scientific Education, Pharmacovigilance, Clinical Planning and Case Ethical Disclosures.
MMS Holdings is a Contract Research Organization that fulfils compulsory steps to help Pharmaceutical companies apply for introducing new drugs into the market. Check them out to know more.
What is the difference between clinical research and medical research?
Clinical research is a subcategory of medical research that involves studies with humans or human-related data. Medical research is concentrating on unlocking the basis of medical treatment by using a variety of approaches.
What is Blinding in Clinical Trial?
Blinding is a procedure in which one or more parties in a trial are unaware of which participants in the treatment arms were assigned, that is, what treatment was received.
Blinding is an essential characteristic of any study performed to prevent and eliminate conscious or implicit bias in planning and conducting a clinical trial.
What are the Types of blinding?
A clinical trial is called a single blind if only one group, usually the patients, are blinded. If both the participants and study staff are blinded, it is known as double blind study. Triple blinded experiments also expand the data analysts to blind.
A trial in which no blinding is used and the treatment groups are known to all parties is called open label or unblinded.
| Type | Description |
| Unblinded or open label | All parties are aware of the treatment the participant receives |
| Single blind or single-masked | Only the participant is unaware of the treatment they receive |
| Double blind or double-masked | The participant and the clinicians / data collectors are unaware of the treatment the participant receives |
| Triple blind | Participant, clinicians / data collectors and outcome adjudicators / data analysts are all unaware of the treatment the participant receives |


 Update: Daily Roundup July 14, 2020")

?")
")