
As per WHO: “Pharmacovigilance (PV) is defined as the science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other drug-related problem.
The aims of PV are to enhance patient care and patient safety in relation to the use of medicines; and to support public health programmes by providing reliable, balanced information for the effective assessment of the risk-benefit profile of medicines.”
Table of Contents
Introduction
PV is the science of collecting, monitoring, researching, evaluating and analysing evidence from health care providers and patients on the adverse effects of medicinal products, biologics, blood products, herbals, vaccines, medical devices, traditional and complementary medicinal products with a view to identifying new information on product-related hazards and preventing patient harms.
PV’s goals are to enhance patient safety regarding medicinal use by providing a system for collecting, evaluating and distributing data on drug safety. PV activities involve monitoring approved drugs and research medicines (IMPs) to:
• Determine previously unknown adverse reactions
• Detect changes in the frequency or severity of known adverse reactions
• Assess the risk / benefit of drugs to determine if measures are needed to improve safety
• Ensure the accuracy of information communicated to healthcare professionals and patients and ensure up-to – date information contained in PILs.
Why is Pharmacovigilance important?
Pharmacovigilance is key to safety of drugs. PV analyses conducted in clinical trials of Phase I, Phase II and Phase III give drug companies data on the drug’s safety profile. Where necessary, these data may be used for further R&D or may be submitted to regulatory authorities to allow access to new markets.
Both PV practises in clinical research and those performed through medical professionals and consumers offer valuable insights into the pharmaceutical drug safety profile.
When identifying a new adverse reaction it is necessary to update the list of side effects on the label. Due to dangerous side effects, PV data can occasionally lead to the removal of a drug from the market (drug recall).
Scope of Pharmacovigilance
Since the WHO Technical Report in 1972, the PV discipline has developed significantly, and it remains a dynamic clinical and scientific discipline. The challenges of increasing the range and potency of pharmaceutical and biological medicines including vaccines, which carry with them an inevitable and at times unpredictable potential for harm, have been addressed.
On the other hand, the risk of harm is less when medicines are used by an informed health profession and by patients who themselves understand and share responsibility for their drugs.
When adverse effects and toxicity appear, especially when previously unknown in combination with the medicine, it is essential that they are analysed and effectively communicated to an audience that has the knowledge to interpret the information. That is PV ‘s role, much of which has already been achieved.
But it takes more for the discipline to be integrated into clinical practise and public policy. A pharmaceutical company in India must essentially carry out activities such as collection and expedited reporting of serious unexpected ADRs in order to fulfil the PV obligations for its marketed products as per the regulations.
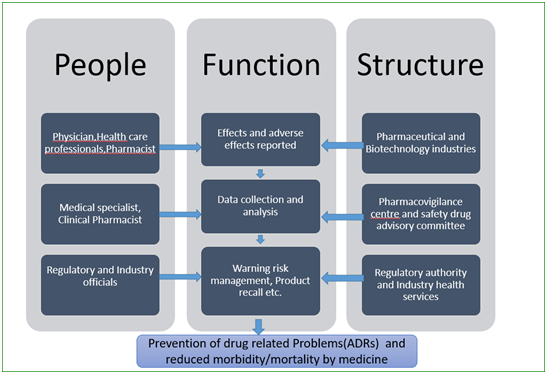
A typical setup for PV studies is shown in Figure below that includes people involved at different levels, organisational units and their functions.
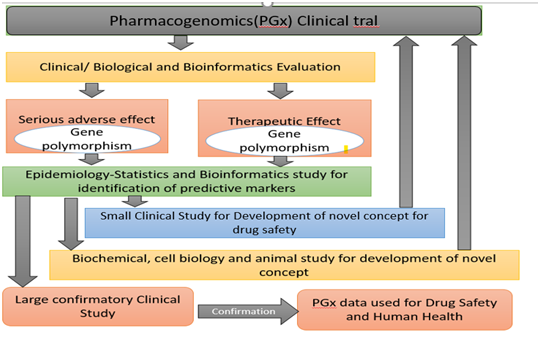
Role of Pharmacogenomics in PV
Pharmacogenomics (PGx) combines traditional pharmaceutical sciences such as biochemistry with annotated knowledge of genes , proteins, and single nucleotides polymorphisms (SNP).
It is the technology that deals with the effects or toxicity of a drug by correlating gene expression or single-nucleotide polymorphisms (SNP) with the influence of genetic variation on drug response in patients.
In doing so, (PGx) aims to develop rational means to optimise drug therapy with regard to the genotype of patients, to ensure maximum effectiveness with minimal adverse effects.
Such approaches promise the advent of “personalised medicine;” in which drugs and combinations of drugs are optimised for the unique genetic makeup of each individual.
The science of pharmacogenetics (PG) originated from the analysis of a few rare and sometimes inadvertently observed extreme reactions (phenotypes) in some people. These phenotypes were either diseases inherited or anomalous drug reactions or other environmental factors.
Research on PG and PGx remain challenged, and there is more room for improvement opportunities in each approach.
Figure 3 shows that multiple approaches need to be combined to acquire knowledge of PGx that is valuable for the development of new therapies or to improve existing therapies.
Pharmacovigilance Risk Assessment Committee
The Pharmacovigilance Risk Assessment Committee (PRAC) of EMA is responsible for evaluating and monitoring the safety of human medicinal products.
It is composed of medicine safety experts nominated by the European Commission from regulatory authorities in Member States , plus scientific experts and representatives of patients and health care professionals.
EMA supports the PRAC by providing data from the clinical practice available in electronic health records or prescription databases.
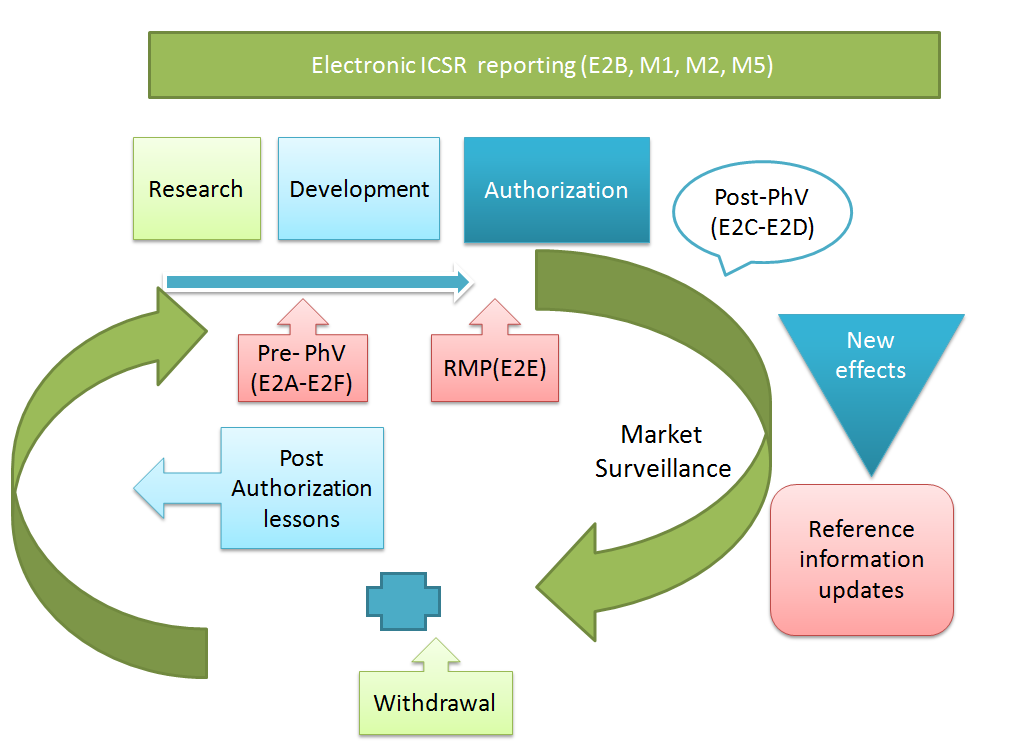
Pharmacovigilance and ICH regulations
Until now, topics related to Pharmacovigilance have entered the ICH process in two waves.
In 1994, the first wave led to the adoption of the ICH Topic ICH-E2A, with an extension of this work in the form of E2B and E2C, finalised between 1996 and 1997. The second wave began in 2002 with three additional ICH topics, E2D, E2C Addendum and E2E, finalised below between 2003 and 2004.
Important points addressed in the ICH-E2A
- Definitions for AE and ADR in the pre-authorization phase.
- Criteria for serious AE/ADR
- Expectedness of an AE/ADR based on clinical observation and its documentation in the applicable product information
- Causality assessment as good case practice for the AE/ADR cases from clinical trials
- Indirect possible causality for spontaneously reported ADR cases
- Standards for expedited reporting from clinical trials
- Definition of minimum case report information for the report submission to authorities
- Follow-up reporting
- Unblinding procedures for serious ADRs
- Reporting of emerging information on post-study ADRs
- Reporting constraint for active comparator
Important points addressed in the ICH-E2D
• The AE and ADR meanings in the post-authorisation process
• Severe AE / ADR requirements according to ICH-E2A;
• Clinical observation and recording of the AE / ADR in the approved product information; class effects explanations
• Distinction between sources of unsolicited and submitted data
• Induced (but not demanded) reporting explanations;
• Increased reporting requirements in post-authorisation process
• Description of minimum case report details to be submitted with explanations to authorities
- Follow-up reporting
- Lack of efficacy reporting needs
• ADR Description Guidelines
• ADR case management guidelines
• Case management of pregnancy exposure
• Explanation of marketing authorisation holder ‘s reporting obligation in view of all contractual relationships in place
Important points addressed in the ICH-E2B(M)
Definition of all ADR case reports data elements: heading and content of data field
• Technical requirements for each data field such as field length and field value and the related additional technical data fields
• List of Unit Abbreviations
• Number of Time Intervals Units
• List of Administration routes
Important points addressed in the ICH-E2C
- Inclusion of all product descriptions in one PSUR (Periodic safety update report)|
- Definition of foreign product birthdates, evaluating the PSUR data lock points
- Provision for the submission of a set of PSURs, covering each subsequent 6 months, to encourage the submission of PSUR at local frequency
- Overview of all sources of data to be included in a PSUR
- Incorporation of worldwide marketing authorisation status information and regulatory safety-related steps, ADR and exposure data
- Use of Company Core Safety Information (CCSI) as a reference and definition for the non-listed ADR (i.e., unlisted compared to CCSI versus unforeseen compared to local product information);
- Overview of case history by case
- Formats of ADR line listings and summary tabulations
- Presentation of the exposure data
Important points addressed in the ICH-E2E
- Safety specification elements as overview of known risks, risks that may emerge from populations and circumstances that have not yet been properly examined, and other possible risks
- Type of a safety relevant pharmacovigilance plan
- The pharmacovigilance plan requires the definition of routine pharmacovigilance as a minimum and the inclusion of a safety action plan for particular concerns / the lack of knowledge as required
- Format of the action plan for protection, outlining the justification for action and the timeline for assessment and reporting (‘milestones’)
- Possible timetable alignment with regulatory timetable for post-authorisation review, such as PSUR review or renewal evaluation of marketing authorizations.
- Principles for designing and conducting non-experimental clinical pharmacoepidemiological trials with references to international guidelines.
- Overview of data collection techniques for evaluating known or unknown hazards and comparison.
Pharmacovigilance in Pediatric population
Pediatric population is defined as between the ages of 0 and 18. Pediatrics have been struggling with few available medications designed especially for children since many age groups.
A lack of clinical trials in this age group is the reason behind reduced availability of medicines.
Pediatrists have no choice but to prescribe it on a “off-label” basis, since these drugs have not been properly tested and/or developed and approved for use in the appropriate community of paediatric ages.
And these health care professionals should be aware of the risk associated with prescribing and delivering such medications to children.
The risk of adverse reactions increases with drug use “off-label” and so regulatory authorities play a significant role in reminding health care providers to report adverse drug reactions and paediatric pharmacovigilance procedure.
Specific issues associated with the paediatric population include lack of clinical trials, under or over dosage, lack of pharmacokinetics and dose-finding studies; growth-induced, developmental disabilities and delayed ADRs.
Health practitioners, parents, pharmaceutical industry, patient associations, national healthcare systems, etc., are numerous stakeholders that play a role in pharmacovigilance.
Different regulatory guidance’s available for pediatric pharmacovigilance are but not limited to:
ICHE2E,EMEA : Guideline on conduct of pharmacovigilance for medicines used by the pediatric population. Guideline on conduct of pharmacovigilance for vaccines.
Points to be considered for the future in pediatric pharmacovigilance are:
- Pediatric population should be taken into account during all phases of the pharmacovigilance cycle
- Promote ADR reporting
- Expanding definition of ADR in order to include off-label, misuse, error.
- Risk management plans
- Signal detecting systems
- Additional monitoring system”
References:
https://www.intechopen.com/books/pharmacovigilance/introductory-chapter-pharmacovigilance
https://www.ema.europa.eu/en/human-regulatory/overview/pharmacovigilance-overview
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4576445/
https://www.who.int/medicines/areas/quality_safety/safety_efficacy/pharmvigi/en/
https://www.technologynetworks.com/drug-discovery/articles/what-is-pharmacovigilance-328154
Wonderful blog! Thanks for the information, Also if anyone is interested for Jobs in Clinical Research do visit the institute which provides Pharmacovigilance Course
Please add the blog link in your website