There are different views on using these two terms- medical writing and safety writing. Hence we tried to put some light on this so that you can understand the real difference between scientific writing and medical writing.
Few use them interchangeably as well. We read many online resources on this topic and check the various job descriptions so we will share both understandings here.
Table of Contents
Understanding 1: In many online resources:
So generally scientific writing is considered a big umbrella and Medical writing is a part of it.
So with this logic, all the medical writers are scientific writers, but all the scientific writers are not medical writers.
Medical writing is a much more specific field of scientific writing. Medical writing deals with the development of standard, comprehensive, reliable, skilful, and convincing clinical and scientific documentation.
Medical writing is an art of writing more efficiently and clearly about a particular content that can be simply understood as a target audience.
And all the writing-related to science can be considered under scientific writing categories. It may be medical journalism, Protocol writing. PSUR, IND, Manuscript etc. see the below NCBI article:
So organisations/employers use medical writer designation especially in their job description if they are seeking candidates for writing below documents.
Package Inserts (prescribing information) & Patient Information Leaflets
Subject narratives
and other regulatory documents
You can see with the above examples, that medical writers are involved in writing exclusively medical and health-related documents and these documents follow country and organization-specific format and regulatory guidelines.
So Medical writing jobs require a good domain and regulatory guidelines knowledge along with excellent writing skills.
And all the writings except the above documents, fall under scientific writing categories.
Medical Journalism
Clinical trial publication
Press release
Medical education
Website content writing on health, clinical or other
August 20, 2020: “The U.S. Food and Drug Administration is providing an update on adverse events reported to the Agency related to breast implants, including breast implant-associated anaplastic large cell lymphoma (BIA-ALCL), and systemic signs and symptoms referred to by patients as breast implant illness (BII), which some patients report after receiving breast implants.
The FDA is also qualifying the BREAST-Q Reconstruction Module as a medical device development tool (MDDT) to aid in the assessment of certain medical devices such as breast implants. Qualification of the BREAST-Q Reconstruction Module MDDT included the Physical Well-being (Chest), Psychosocial Well-being, Sexual Well-being and Satisfaction with Breasts scales.
An MDDT is scientifically validated and can be qualified for use in device evaluation and to support regulatory decision-making. Examples of MDDTs are clinical outcome assessments, assessments of biomarkers, and non-clinical assessment methods or models. The use of a qualified MDDT by a product sponsor is voluntary.
“The FDA has been diligently monitoring adverse events associated with breast implants for decades and has been working to better understand the quality of life and satisfaction a breast reconstruction patient may experience in order to refine our evaluation of breast implant benefits and risks.
Our qualification of the BREAST-Q Reconstruction Module as a validated tool to assess outcomes of breast reconstruction surgery in terms of quality of life and satisfaction helps accomplish this,” said Binita Ashar, M.D., director of the Office of Surgical and Infection Control Devices in the Center for Devices and Radiological Health.
“In addition, we continue to increase our scientific knowledge regarding BIA-ALCL and systemic symptoms referred to as BII, and remain committed to keeping the public informed.”
Medical Device Reports of BIA-ALCL
The FDA analysis of global medical device reports for BIA-ALCL covers reports received through January 5, 2020. It updates the FDA’s last public report with new information from July 7, 2019 to January 5, 2020.
The FDA updated the table on the agency’s BIA-ALCL webpage to include a total of 733 unique cases and 36 patient deaths globally, which reflect an increase of 160 new cases and 3 deaths since the early-July, 2019 update.
Specifically, of the 733 total unique cases of BIA-ALCL reported to FDA, 620 cases were reported for Allergan implants, and 47 cases involved implants with an unknown manufacturer.
With respect to implant surface for the 733 total unique cases of BIA-ALCL, 496 cases were reported to have textured implants, and 209 cases did not specify the implant surface.
Of the 36 total patient deaths reported to FDA, 15 of the 16 patients for which the manufacturer of the implant is known, are reported to have had an Allergan breast implant at the time of their BIA-ALCL diagnosis.
In terms of implant surface, of the 36 cases reported of patient deaths, 16 cases reported textured implants, and 19 cases did not contain information on the implant surface.
BIA-ALCL is not breast cancer—it is a type of non-Hodgkin’s lymphoma (cancer of the immune system). In most cases, BIA-ALCL is found in the scar tissue and fluid near the implant, but in some cases, it can spread throughout the body.
At this time, the overall incidence of developing BIA-ALCL is low; however, a BIA-ALCL diagnosis is serious and can lead to death, especially if not diagnosed early or promptly treated.
In most patients, BIA-ALCL is treated successfully with surgery to remove the implant and the scar tissue surrounding the implant; however, some patients may require treatment with chemotherapy and/or radiation therapy.
Medical Device Reports of Systemic Symptoms Referred to as BII
In addition to today’s update on BIA-ALCL, the FDA is updating data on medical device reports received concerning systemic signs and symptoms referred to by patients as BII.
A new table on FDA’s website summarizes unique BII medical device reports from the U.S. and worldwide that the FDA has received from Jan. 1, 2008 to Oct. 31, 2019.
The data show that the FDA received 2,497 medical device reports containing symptoms consistent with BII from Nov. 2018 to Oct. 2019. The FDA’s data from Jan. 2008 to Oct. 2018 showed 1,080 reports that contained such symptoms.
More patients and providers are reporting these conditions, likely due to increased awareness from press, social media, and FDA’s General and Plastic Surgery Devices Advisory Committee meeting held in March 2019.
While there is limited use of the term “breast implant illness” in medical literature, symptoms such as fatigue, memory loss, rash, “brain fog,” and joint pain may be associated with breast implants, and some patients and clinicians may use the term “breast implant illness” to describe these symptoms or use these terms when reporting them to the FDA.
The top 10 most common symptoms reported to the FDA’s medical device report database for patients with breast implants include fatigue (49 percent), brain fog (25 percent), joint pain (25 percent), anxiety (24 percent), hair loss (21 percent), depression (19 percent), rash (18 percent), autoimmune diseases (18 percent), inflammation (18 percent) and/or weight problems (18 percent). Researchers are investigating these symptoms to better understand their origins and connection to breast implants.
While the FDA doesn’t have definitive evidence demonstrating breast implants cause these symptoms, the current evidence supports that some patients experience systemic symptoms that may resolve when their breast implants are removed.
The FDA is committed to communicating information the agency receives about systemic symptoms reported by patients with breast implants.
Qualification of the BREAST-Q Reconstruction Module as a Medical Device Development Tool
The FDA also announced today the qualification of a validated, self-administered questionnaire—the BREAST-Q Reconstruction Module—through the agency’s MDDT program.
The paper and electronic self-administered versions of the BREAST-Q Reconstruction Module’s Psychosocial Well-being, Sexual Well-being, Physical Well-being (Chest), and Satisfaction with Breasts scales are used to quantify different aspects of a woman’s quality of life and satisfaction with breast reconstruction surgery.
These scales may be used by medical device sponsors and sponsor-investigators in feasibility, pivotal, and post-approval studies to support the effectiveness of breast reconstruction-related medical devices, such as an implant or mesh, befitting the clinical meaningfulness of the scale to support the proposed indication.
The FDA remains committed to thoughtful, scientific and transparent public dialogue concerning breast implant safety and effectiveness.
Health care professionals and consumers should report any adverse events related to breast implants to the FDA’s Adverse Event Reporting Program. The FDA monitors these reports and takes the appropriate action necessary to help ensure the safety of medical products in the marketplace.
Lastly, today the FDA released a video on seven things patients should know about breast implants, including risks, complications and information about BIA-ALCL and systemic symptoms.
The FDA will continue to analyze all available information regarding risks associated with breast implants, routinely update the BIA-ALCL and BII analysis published on our website and take additional actions when and where necessary.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices.
Aug. 19, 2020: “Verona Pharma, a clinical-stage biopharmaceutical company focused on respiratory diseases, announces the initiation of the second, multiple dose, part of a Phase 2 trial to evaluate the pressurized metered-dose inhaler (“pMDI”) formulation of ensifentrine in patients with moderate to severe chronic obstructive pulmonary disease (“COPD”).
Results from the study (Part B) are expected in the first half of 2021.
Positive efficacy and safety data from the first, single dose, part of the study (Part A) in 40 patients with moderate to severe COPD were announced by the Company on March 31, 2020.
The results demonstrated a statistically significant and clinically meaningful increase in lung function as measured by forced expiratory volume in one second (“FEV1”) compared to placebo.
Verona Pharma decided to postpone initiation of Part B of the study due to concerns for the safety of patients and study staff because of the COVID-19 pandemic.
Following an assessment of local infection rates and control measures in addition to procedures put in place by the UK clinical sites, the Company has now initiated Part B, which will evaluate the pMDI formulation.
Multiple Dose Crossover Trial, Part B
Patient Population: Approximately 30 moderate to severe COPD patients who participated in Part A are planned to continue to Part B at two sites in the UK.
Dose/Duration: Patients will be randomized to receive 3 dose levels (300 µg, 1000 µg, 3000 µg) of pMDI ensifentrine or placebo, twice-daily over one week. All patients will receive each of the dose levels and placebo over four 7-day treatment periods.
Primary Endpoint: Improvement in lung function as measured by peak FEV1 with ensifentrine compared to placebo after 7 days of treatment.
Secondary Endpoints: Safety and tolerability, other lung function measures such as trough FEV1, average FEV1 over 4 and 12 hours, and steady state pharmacokinetic profile of ensifentrine pMDI.
“We are pleased to start the multiple dose part of this pMDI study and expect the results in the first half of 2021,” said David Zaccardelli, Pharm. D., President and CEO of Verona Pharma.
“Data from the single dose part of this pMDI study are very encouraging and consistent with data from Phase 2 clinical trials with our nebulized and dry powder inhaler (“DPI”) formulations of ensifentrine.
pMDI and DPI formulations are important delivery mechanisms in the approximately $9.6 billion US market for maintenance COPD therapies.
The development of pMDI and DPI formulations of ensifentrine provides expanded opportunities including life cycle management, new indications and partnering.
August 17, 2020: “The U.S. FDA has approved Enspryng (satralizumab-mwge) for the treatment of neuromyelitis optica spectrum disorder (NMOSD) in adults with a particular antibody – patients who are anti-aquaporin-4 or AQP4 antibody-positive.
NMOSD is a rare autoimmune disease of the central nervous system that mainly affects the optic nerves and spinal cord. Enspryng is the third approved treatment for the disorder.
“Until last year, there were no FDA-approved treatments for patients with this rare, debilitating and sometimes fatal disease. Now there are three,” said Billy Dunn, M.D., director of the Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research.
“Today’s approval of Enspryng highlights the FDA’s commitment to rapidly advancing safe and effective therapies for NMOSD and other neurological diseases.”
In patients with NMOSD, the body’s immune system mistakenly attacks healthy cells and proteins in the body, most often those in the optic nerves and spinal cord.
Individuals with NMOSD typically have attacks of optic neuritis, which causes eye pain and vision loss. Approximately 50% of patients with NMOSD have permanent visual impairment and paralysis caused by NMOSD attacks.
Estimates vary, but NMOSD is thought to impact approximately 4,000 to 8,000 Americans.
NMOSD can be associated with antibodies that bind to a protein called aquaporin-4 (AQP4). Binding of the anti-AQP4 antibody appears to activate other components of the immune system, causing inflammation and damage to the central nervous system.
The effectiveness and safety of Enspryng for the treatment of NMOSD was demonstrated in two 96-week clinical studies.
The first study included 95 adult patients; 64 of these patients had antibodies against AQP4 (anti-AQP4 positive). During this study, treatment with Enspryng reduced the number of NMOSD relapses by 74% in patients who were anti-AQP4 positive compared to treatment with a placebo (inactive treatment).
The second study included 76 adult patients; 52 of these patients were anti-AQP4 positive. During the second study, treatment with Enspryng reduced the number of relapses in patients who were anti-AQP4 positive by 78% compared to treatment with a placebo.
There was no evidence of a benefit in patients who were anti-AQP4 antibody negative in either trial.
The prescribing information for Enspryng includes a warning for increased risk of infection, including serious and potentially fatal infections – such as potential reactivation of hepatitis B and tuberculosis.
Other warnings and precautions for Enspryng include elevated liver enzymes, decreased neutrophil counts and hypersensitivity reactions.
The most common side effects observed were the common cold (nasopharyngitis), headache, upper respiratory tract infection, inflammation of the lining of the stomach, rash, joint pain, extremity pain, fatigue and nausea.
Vaccination with live-attenuated or live vaccines is not recommended during treatment and should be administered at least four weeks before starting Enspryng.
Enspryng received fast track designation, which expedites the development and review of drugs that are intended to treat a serious condition and demonstrate the potential to address an unmet medical need. The drug also received orphan drug designation, which provides incentives to assist and encourage drug development for rare diseases.
The FDA is granting the approval to Genentech Inc.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices.
August 17, 2020: Roche announced today that the US FDA has approved ENSPRYNG™ (satralizumab-mwge) as the first and only subcutaneous treatment for adults living with anti-aquaporin-4 (AQP4) antibody positive neuromyelitis optica spectrum disorder (NMOSD).
NMOSD is a rare, lifelong and debilitating autoimmune disorder of the central nervous system, often misdiagnosed as multiple sclerosis, that primarily damages the optic nerve(s) and spinal cord, causing blindness, muscle weakness and paralysis.
“Today’s FDA approval of ENSPRYNG, the first subcutaneous NMOSD treatment using novel recycling antibody technology, builds upon the work we’ve done in multiple sclerosis with OCREVUS to develop first-in-class medicines and further the scientific understanding of neuroimmunological diseases,” said Levi Garraway, M.D., Ph.D., Chief Medical Officer and Head of Global Product Development. “We thank the NMOSD community, including patients and investigators who participated in ENSPRYNG clinical trials.”
ENSPRYNG is a humanised monoclonal antibody and the only approved therapy designed to target and inhibit interleukin-6 (IL-6) receptor activity, believed to play a key role in the inflammation associated with NMOSD.
The treatment was designed by Chugai, a member of the Roche group, using novel recycling antibody technology, which compared to conventional technology, allows for longer duration of antibody circulation and subcutaneous dosing every four weeks.
“For people with NMOSD, relapses can cause devastating, irreversible and disabling neurological effects,” said Professor Jeffrey Bennett, University of Colorado Neurology & Ophthalmology, and investigator for the ENSPRYNG pivotal clinical trials.
“Having an approved therapy that can be administered subcutaneously in the home, and has demonstrated an impact on the frequency of relapses, is an important advancement for patients.”
“We are very optimistic the addition of this new approved treatment option will make a meaningful difference for those living with NMOSD, those who love and support them and the doctors who treat them,” said Victoria Jackson, founder, The Guthy-Jackson Charitable Foundation.
“When my daughter was diagnosed with NMOSD in 2008, there were no approved treatment options, and a critical lack of resources and understanding for people living with this disabling disorder.
After years of dedicated effort and collaboration, the FDA approval of ENSPRYNG exemplifies how patients, industry, and academia can find solutions together.”
ENSPRYNG can be administered in the home by a person living with NMOSD or a caregiver following training from a healthcare provider. ENSPRYNG treatment is administered every four weeks after an initial loading dose.
ENSPRYNG will be available in the United States in two weeks.
FDA approval is based on results from one of the largest pivotal clinical trial programmes undertaken for this rare neurological disorder
This approval is supported by results from two randomised controlled Phase III clinical trials, the SAkuraStar and SAkuraSky studies, in which ENSPRYNG demonstrated robust and sustained efficacy and a favourable safety profile in adults with AQP4 antibody positive NMOSD.
Results were sustained for 96 weeks, significantly reducing the risk of relapse compared with placebo as a monotherapy and when used concurrently with baseline immunosuppressant therapy (IST), which has commonly been used to manage NMOSD symptoms associated with relapses.
In the SAkuraStar monotherapy study’s AQP4 antibody positive subgroup, 76.5% of ENSPRYNG-treated patients were relapse-free at 96 weeks, compared to 41.1% with placebo.
In the SAkuraSky study, which evaluated ENSPRYNG when used concurrently with baseline IST, 91.1% of ENSPRYNG-treated AQP4 antibody positive subgroup patients were relapse-free at 96 weeks, compared to 56.8% with placebo. The primary endpoint of both SAkuraStar and SAkuraSky was time to first protocol-defined relapse (PDR) adjudicated by an independent review committee in the double-blind period.
The most common adverse reactions with ENSPRYNG (incidence ≥ 15%) were nasopharyngitis, headache, upper respiratory tract infection, gastritis, rash, arthralgia, extremity pain, fatigue and nausea. https://www.roche.com/media/releases/med-cor-2020-08-17.htm
August 18, 2020: AstraZeneca’s Imfinzi (durvalumab) has received acceptance for its supplemental Biologics License Application (sBLA) and has also been granted Priority Review in the US for a new four-week, fixed-dose regimen for treatment in the approved indications of non-small cell lung cancer (NSCLC) and bladder cancer.
If approved, Imfinzi could be administered intravenously every four weeks at a fixed dose of 1500mg in unresectable Stage III NSCLC after chemoradiation therapy and previously treated advanced bladder cancer, consistent with the approved dosing in extensive-stage small cell lung cancer (ES-SCLC).
If approved, the new dosing will be available as an alternative to the approved weight-based dosing of 10mg/kg every two weeks. The sBLA was based on data from several Imfinzi clinical trials, including results from the Phase III CASPIAN trial in ES-SCLC which used the four-week, fixed-dose regimen during maintenance.
Dave Fredrickson, Executive Vice President, Oncology Business Unit, said: “The new less-frequent dosing option for non-small cell lung cancer and bladder cancer will simplify and improve treatment by enabling continuity of care while minimising the risk of exposure to infection in the healthcare setting.
This takes on particular urgency during the current pandemic, as doctors care for patients at high risk of COVID-19 complications. We are working with health authorities in the US and other countries to bring the option of four-week, fixed dosing for Imfinzi to patients around the world as soon as we can.”
Imfinzi is approved in the curative-intent setting of unresectable, Stage III NSCLC after chemoradiation therapy in the US, Japan, China, across the EU and in many other countries, based on the Phase III PACIFIC trial.
Imfinzi is also approved for previously treated patients with advanced bladder cancer in the US and several other countries. Additionally, it is approved in the US and under review in Japan and other countries for ES-SCLC.
Imfinzi was also recently recommended for marketing authorisation in the EU for this indication.
Stage III NSCLC
Stage III (locally-advanced) NSCLC is commonly divided into three sub-categories (IIIA, IIIB and IIIC), defined by how much the cancer has spread locally and the possibility of surgery.
Stage III disease is different from Stage IV disease, when the cancer has spread (metastasised), as the majority of Stage III patients are currently treated with curative intent.
Stage III NSCLC represents approximately one-third of NSCLC incidence and in 2015 was estimated to affect nearly 200,000 patients in the following eight key countries: China, France, Germany, Italy, Japan, Spain, UK, and the US, with approximately 43,000 cases in the US alone. The majority of Stage III NSCLC patients are diagnosed with unresectable tumours.
August 17, 2020: “The U.S. Food and Drug Administration announced the following actions taken in its ongoing response effort to the COVID-19 pandemic:
The FDA issued Yale School of Public Health an emergency use authorization (EUA) for its SalivaDirect COVID-19 diagnostic test, which uses a new method of processing saliva samples when testing for COVID-19.
This molecular test is for the qualitative detection of nucleic acid from SARS-CoV-2 in saliva collected without preservatives in a sterile container from individuals suspected of COVID-19 by their healthcare provider.
SalivaDirect does not require any special type of swab or collection device – a saliva sample can be collected in any sterile container. It is also unique because it does not require a separate nucleic acid extraction step. This is significant because the extraction kits used for this step in other tests have been prone to shortages in the past.
As part of the FDA’s effort to protect consumers, the agency issued a joint warning letter with the Federal Trade Commission to SilveryGuy, a company that participates in the Amazon Associates program, for selling fraudulent COVID-19 related products.
As an Amazon associate, the company earns commissions by promoting the sale of Colloidal Silver products on the company’s website, with misleading claims that the product can mitigate, prevent, treat, diagnose, or cure COVID-19 in people.
Currently, there are no FDA-approved products to prevent or treat COVID-19. Consumers concerned about COVID-19 should consult with their health care provider.
To date, the FDA has currently authorized 214 tests under EUAs; these include 175 molecular tests, 37 antibody tests, and 2 antigen tests.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices.
According to the National Cancer Institute, a biomarker is “a biological molecule found in blood, other body fluids, or tissues that is a sign of a normal or abnormal process, or of a condition or disease,”(NCI) such as cancer. Biomarkers typically differentiate an affected patient from a person without the disease.
Table of Contents
What is biomarker?
“A biomarker — short for “biological marker”—is anything that can be assessed objectively and is either a sign of a normal or abnormal procedure, or a disease or illness. A biomarker may be a molecule present in the blood, or other tissue or body fluids.
Another form of biomarker is a genetic signature or “fingerprint” — an activity pattern in a collection of genes that indicates a certain biological condition.
Examples of biomarkers cover everything from pulses and blood pressure to simple chemistries to more complicated blood and other tissue tests in the laboratory.
In oncology, a biomarker may be a molecule that is secreted by a tumor, or a body ‘s specific reaction to cancer.
This may help to detect cancers in the early stages, forecast how dangerous a cancer can be or predict how well a patient may respond to treatment.
Some examples of biomarkers used for cancer screening or treatment effectiveness tests include AFP (liver cancer), BCR-ABL (chronic myeloid leukemia), CA-125 (ovarian cancer), CA19.9 (pancreatic cancer) and CEA (colorectal cancer).
Biomarkers also help to predict or control recurrence of cancer. For example, the Oncotype DX ® test on breast cancer is used to determine the risk of recurrence of breast cancer in women being treated for early breast cancer.
Oncotype DX tests a set of 21 genes in cells taken during tumor biopsy, and offers a risk score for recurrence.
Additionally, cancer biomarkers have demonstrated usefulness in measuring how well a drug performs over time. Researchers continue to explore the potential of these biomarkers as alternatives to current image based research, such as CT scans and MRIs.
One of biomarker research’s strongest fields is immunotherapy, a form of treatment that manipulates a patient’s immune system to combat drug cancer or modified immune T cells.
In recent years, immunotherapy has had significant success in certain patients with some forms of cancer, but as of now, only a minority of patients are responding to treatment with immunotherapy agents.
Researchers are working hard to find biomarkers that could identify, which patients are likely to respond to immunotherapy in advance. In some forms of cancer, a better or worse response to immunotherapy was associated with the presence or absence of immune “checkpoint” molecules, such as PD1 on immune cells or PDL-1 in cancer cells.
For example, one clinical trial showed that the immunotherapy drug pembrolizumab lowered the risk of death in patients with non-small cell lung cancer – and whose tumors expressed high levels of PDL-1—by 37 percent compared to chemotherapy.
Molecular – have biophysical properties, which allow their measurements in biological samples Eg, Blood glucose plasma, serum, cerebrospinal fluid, bronchoalveolar lavage, biopsy
Radiographic – obtained from imaging studies
Eg: Bone mineral density
Histologic – reflect biochemical or molecular alteration in cells, tissues or fluids
Eg: Grading and staging of cancers
Physiologic – measures of body processes
Eg: Blood pressure
Biomarkers can indicate a variety of health or disease characteristics, including the level or type of exposure to an environmental factor, genetic susceptibility, genetic response to environmental exposure, markers of subclinical or clinical disease, or indicators of response to the therapy.
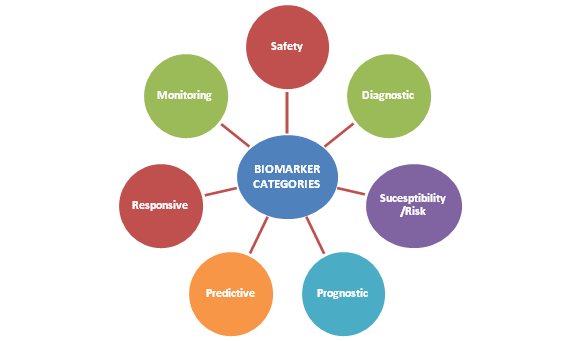
Applications of Biomarkers
Biomarkers have many useful applications in health care, including prevention and diagnosis of diseases, risk assessment for specific diseases, and monitoring of diseases. They may also be used to assess the safety or toxicity of a treatment procedure, or other exposures to the environment.
Applications of Biomarkers
Biomarker Application
Biomarker Examples
Disease state
Screening
Prostate specific antigen (PSA)
Prostate cancer
Fecal occult blood test
Colon cancer
Susceptibility/Risk
Breast cancer genes 1 and 2 mutations (BRCA 1/2)
Predisposition to developing breast cancer
Factor V Leiden
Predisposition to developing thromboembolism
Apolipoprotein C
Predisposition to developing Alzheimer’s disease
Human papillomavirus (HPV)
Predisposition to developing cervical cancer
Diagnostic
Troponin-I
Coronary ischemia
Sweat chloride
Cystic fibrosis
Ejection fraction (EF)
Cardiomyopathy/congestive heart failure
Glomerular filtration rate (GFR)
Chronic kidney disease
Prognostic
BRCA ½
Likelihood of second cancer in women with breast cancer
Chromosome 17p deletions and TP53 mutations
Likelihood of death in patients with chronic lymphocytic leukemia
Monitoring
Serum low-density lipoprotein (LDL)
Response to lipid lowering drugs
International Normalized Ratio (INR)
Efficacy of anticoagulant therapy
Cancer antigen 125 (CA-125)
Ovarian cancer disease status or burden
Hemoglobin A1C
Response to antihyperglycemic agents or lifestyle changes
Viral load
Response to antiretroviral treatment for human immunodeficiency virus (HIV)
Safety
Hepatic aminotransferases and Bilirubin
Hepatotoxicity
Serum creatinine
Nephrotoxicity
Serum potassium
Hypo or Hyperkalemia while taking diuretics, angiotensin converting enzyme inhibitors
Corrected QT interval (QTc)
Assess potential for drugs to induce ventricular tachycardia
Serum Bisphenol A (BPA)
Measured in pregnant women; predicts adverse birth outcomes such as preterm birth; has informed/improved public safety through BPA-free plastics.
The use of validated biomarkers has become widespread both in basic and clinical science and in clinical practice, and their use as endpoints in clinical trials is now generally recognized.
Biomarkers allow for better understanding disease processes and the ways in which drugs work to fight disease. This information may be used earlier to treat, or to avoid, illness before it starts.
Biomarkers may be used to boost the effectiveness and safety of current drugs, and to produce new drugs. New molecular biomarkers have the ability to tailor disease prevention and treatment, making healthcare delivery more reliable, safe and cost-effective and enhancing patient outcomes.”
August 17, 2020: “Sanofi and Principia Biopharma, a late-stage biopharmaceutical company focused on developing treatments for immune-mediated diseases, entered into a definitive agreement under which Sanofi will acquire all of the outstanding shares of Principia for $100 per share in cash, which represents an aggregate equity value of approximately $3.68 billion (on a fully diluted basis).
The Sanofi and Principia Boards of Directors unanimously approved the transaction.
“This acquisition advances our ongoing R&D transformation to accelerate development of the most promising medicines that will address significant patient needs,” said Paul Hudson, Sanofi Chief Executive Officer.
“The addition of multiple BTK inhibitors to our pipeline demonstrates our commitment to strategic product acquisitions in our priority therapeutic areas. Full ownership of our brain-penetrant BTK inhibitor ‘168 removes complexities for this priority development program and simplifies future commercialization.”
“The Phase 2b data in relapsing multiple sclerosis showed the strong potential of ‘168 to address disability and disease progression, and triggered the start of Phase 3 studies across the full spectrum of MS.
Through this acquisition, we will be able to expand and accelerate development of BTK inhibitors across multiple indications. Both ‘168 and rilzabrutinib, have ‘pipeline in a product’ potential, and we look forward to unlocking their full treatment benefits across an array of diseases,” said John Reed, M.D., Ph.D., Global Head of Research & Development at Sanofi.
“Principia’s successful design and development of a whole portfolio of BTK inhibitors for immunology is aimed to transform the treatment for patients with immune-mediated diseases.
By combining with Sanofi, we will bring significant resources to expand and accelerate the potential benefits of these therapies. The benefit of developing several BTK inhibitors will allow us to target specific organ systems for optimal patient benefit.
The merger will provide global resources to get these novel therapies to patients faster,” said Martin Babler, President and CEO at Principia Biopharma.
Principia’s Bruton tyrosine kinase (BTK) inhibitors add to Sanofi’s efforts to accelerate and build a portfolio of the next generation of transformative treatments for autoimmune diseases.
BTK is present in the signaling pathways of key innate and adaptive cell types of the immune system. Being able to block or disrupt these signaling processes can help in stopping inflammation and tissue destruction related to autoimmune diseases and target some of the underlying pathophysiology.
BTK inhibitor ‘168: In a Phase 2b study in patients with multiple sclerosis, ‘168 reduced Gd-enhancing T1 hyperintense lesions by 85% compared to placebo.
In June, Sanofi announced the first multiple sclerosis patient was enrolled in the Phase 3 program for the BTK inhibitor, comprising four pivotal clinical trials across the disease spectrum.
The Principia acquisition will provide an opportunity to expand the development program to evaluate indications beyond central nervous system diseases.
Rilzabrutinib: This oral BTK inhibitor is currently being evaluated in a Phase 3 program for patients with moderate to severe pemphigus, a rare, debilitating autoimmune disease that causes blistering of the skin and mucous membranes.
A Phase 3 program for immune thrombocytopenia, a disease that causes high risk for bleeding events, is expected to be initiated by the end of 2020, assuming no COVID-19 related impact.
The company also has an ongoing Phase 2 program for IgG4-related diseases, which is driven by chronic inflammation, immune cell infiltration, and fibrosis within organs that can lead to severe morbidity.
PRN473 Topical: This BTK inhibitor is a topical agent currently in Phase 1 trials and is being developed for immune-mediated diseases that could benefit from localized application to the skin.
The Principia BTK inhibitor franchise is based on its proprietary Tailored Covalency® platform that has generated potential best-in-class clinical candidates.
The platform allows the design of both reversible covalent and irreversible covalent small molecule inhibitors that are more selective with less off-target effects.
The optimized target residence time has potential to deliver a desired efficacy with a stronger safety profile.
In 2017, Sanofi formed a collaboration with Principia under which Principia granted Sanofi an exclusive, worldwide license to develop and commercialize BTK inhibitor ‘168 in multiple sclerosis and other central nervous system diseases.
Transaction Terms
Under the terms of the merger agreement, Sanofi will commence a cash tender offer to acquire all outstanding shares of Principia common stock for $100 per share in cash for a total enterprise value of approximately $3.36 billion.
The consummation of the tender offer is subject to customary closing conditions, including the tender of at least a majority of the outstanding shares of Principia common stock, the expiration or termination of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, and other customary conditions.
Following the successful completion of the tender offer, a wholly owned subsidiary of Sanofi will merge with Principia and the outstanding Principia shares not tendered in the tender offer will be converted into the right to receive the same $100 per share in cash paid in the tender offer.
The tender offer is expected to commence later this month. Sanofi plans to finance the transaction with cash on hand. Subject to the satisfaction or waiver of customary closing conditions, Sanofi expects to complete the acquisition in the fourth quarter of 2020.
Evercore is acting as financial advisor to Sanofi and Weil, Gotshal & Manges LLP is acting as its legal counsel. Centerview Partners LLC and BofA Securities are acting as financial advisors to Principia and Cooley LLP is acting as its legal counsel.” https://www.sanofi.com/en/media-room/press-releases/2020/2020-08-17-07-00-00
“The Institutional Review Board (IRB) also known as an Independent Ethics Committee (IEC), Ethical Review Board (ERB), or Research Ethics Board (REB), is a kind of committee that applies research ethics by evaluating the methods used for research to ensure that they are ethical.
These boards are formally designated for the acceptance (or rejection), monitoring and review of human biomedical and behavioral studies.
Table of Contents
What is Institutional Review Board?
The Institutional Review Board ( IRB) is an administrative body set up to protect the rights and health of human research subjects recruited to participate in research activities carried out under the auspices of the institution it is associated with.
They also perform some form of risk-benefit analysis in an attempt to decide whether research should be carried out or not.
The goal of the IRB is to ensure that adequate measures are taken to protect the rights and welfare of the participating human beings as subjects in a research study.
Most developing countries, along with developed countries, have established national, regional, or local IRBs to safeguard ethical conduct of research on national and international norms, laws, or codes.
The IRB is concerned with safeguarding human subjects’ health, freedoms and privacy.
The IRB has the authority to approve, oppose, track and mandate changes in all research activities falling under its jurisdiction as defined by both the federal regulations and institutional policies.
IRB Committee Membership
Composition of IRB
Each IRB Committee will be composed of at least five members with varying experience and expertise to provide a thorough and comprehensive review of the institution’s common research activities.
IRB membership must be sufficiently eligible through its members ‘experience and skills and the diversity of its members, including consideration of race, gender, and cultural backgrounds and responsiveness to issues such as community attitudes, to promote respect for its advice and advice in safeguarding human research subjects’ rights and welfare.
Each IRB committee includes persons able to establish the acceptability of the proposed research in terms of institutional commitments and regulations, applicable law, and standards of professional conduct and practice.
Each IRB committee includes members of more than one profession.
Each IRB committee includes at least one member representing the viewpoint of the participants in the research.
Each IRB committee includes at least one member whose primary concern is in scientific areas, and at least one member whose primary concern is in non-scientific areas.
Each IRB committee includes at least one member who is not otherwise affiliated with the University of Pittsburgh, UPMC, CHP, UPMC Cancer Centers, or Magee, and who is not part of the immediate family of a person with such affiliation (i.e., “unaffiliated member”).
Regardless of the level of risk associated with the protocol, research organized by the national Institute for Disability, Independent Living and Recovery Research that specifically includes the inclusion of children with disabilities or people with mental disabilities must be examined by at least one individual who is primarily concerned with the welfare of these research subjects.
This representative must have the necessary scientific or academic expertise to serve in that role. If this is a full board review a committee member may act as a primary or secondary reviewer in this capacity.
In the absence of a suitable reviewer, the IRB may appoint a consultant to act in this position. This will be recorded in Full Board RRC generated minutes.
If the study meets an expedited review category, an suitable consultant will review the protocol. This will be documented on the Documentation Form generated by the EERRC.
Appointment of IRB Members
Institutional Official (IO) shall make appointments of voting members of the IRB Committee. Recommendations for board members may be provided by either the IRB Chair or Associate Director to the IO on the basis of the IRB Committee’s specific needs.
The IRB Chair or his nominee requests recommendations from the Division Chiefs and Department Chairs for faculty volunteers as needed based on factors including, but not limited to, the size, skills and experience needed of the committee; knowledge of the interest of the individual; recommendations of institutional leadership; and/or investigators involved in research studies currently or previously approved by the IRB.
The IRB Chair or his designee reviews the membership rosters and recommends appointment by the Institutional Official of potential non-scientific and/or non-affiliated members to the IRB based on the considerations including, but not limited to: required committee composition, expertise and experience, knowledge of the individual’s interest, recommendations of current or past non-scientific and/or non-affiliated members, and individuals recruited from disease-related organizations or groups.
The IRB Chair and Associate Director must review the CV and demographic sheet for each new member on the IRB rosters for educational background, work history, and current vocation to assess the status of the member (i.e., scientific versus non-scientific, associated versus non-affiliated)
Term of Service
Committee members are primarily appointed to a term of three years. Committee members may be requested to accept reappointment to the IRB for an additional term of three years at the caution of the Chair.
A determination regarding an additional reappointment period will be made at the end of the six-year term. If a member declines full membership, he / she may be asked to become a member alternate.
An updated CV and demographic sheet will be requested from the reappointed members.
Compensation of IRB Members
Representatives of the associated IRB Committee will not receive any direct monetary compensation for membership in the body. Unaffiliated members of the IRB Committee will be reimbursed for internet access at an rate not to exceed $50 a month. Refund payments are to be made on a quarterly basis.
Indemnification
University Policy 07-06-06 sets forth the conditions under which indemnification and legal defense may be available to faculty and staff.
University Policy 07-06-07 sets forth the conditions under which indemnification and legal defense may be available to volunteers at the University. Indemnification may be afforded to the IRB members as set forth in these policies.
Responsibilities of IRB Members
General Responsibilities of all IRB Members include:
Reviewing research study proposals and then evaluating them from the perspective of the regulatory criteria for approval addressed under 45 CFR 46.111, 21 CFR 56.111 (if applicable); and any other relevant ethical, scientific or compliance considerations;
Reviewing and evaluating informed consent documents from the perspective of addressing the necessary and additional elements of informed consent described under 45 CFR 46.116, 21 CFR 50.20 (if applicable) and any other related ethical or regulatory considerations;
Attending at least 70 percent of IRB meetings in person, unless challenging circumstances prevent such attendance from occasional occasions, reporting promptly at the time specified to convene the meeting; and remaining at the meeting until the full agenda has been addressed:
Participating in IRB meetings on problems associated in the planned study studies and relevant informed consent documents, and making suggestions for risk management and enhancement of informed consent process and otherwise enhancement of the security of human subjects;
Voting for full approval, approval subject to modification(s), reconsideration, or disapproval of the human subject research as outlined in Chapter;
Evaluating the risk level (i.e., minimal or greater than minimal) of the proposed research. In performing this evaluation, IRB members will use the following absolute definition for “minimal risk” at 45 CFR 46.102(i) unless the research is directed at prisoner-subjects:
“Low risk means that the probability and severity of harm or discomfort expected in the study is not greater in and of itself than those typically experienced in everyday life ( i.e., in the general population) or during regular physical or psychological exams or tests.”
Deciding, for the research studies of greater than minimal risk, if IRB continuing review of the research is justified on a more frequent basis than the requisite annual review. In making this determination, IRB members will follow the procedure outlined in Chapter;
Deciding, where the informed consent process and/or other aspects of the research study should be audited by the ORP Education and Compliance Office of the University of Pittsburgh for research studies involving greater than minimal risk, difficulty or conflict of interest concerns;
Deciding, for the research studies involving an unapproved device, if the device and its proposed use represent a non-significant or significant risk to research subjects;
Deciding, for research studies subject to IRB continuation approval, if verification from sources other than the investigator is needed that there have been no material changes since the previous IRB review using the criteria outlined in Chapter 12;
Recommending improvements to the IRB policies and procedures so as to improve IRB review process and/or human subject protections;
Informing the IRB Chair or an IRB Vice Chair of human subject research noncompliance problems or ethical issues of which they become aware;
Conforming their behavior in compliance with the legal and ethical principles approved by the IRB at all times; including, but not limited to, upholding the confidentiality / non-disclosure of human subject work submitted for review and approval by the IRB, and engaging in good faith discussions by the IRB without indication of discrimination or conflict of interest.
The IRB must have at least five members with varying histories to provide a complete and accurate analysis of human research and its administrative , legal, scientific, and social implications.
The board will also have at least one non-institutionally associated member and one non-scientist Independent review of clinical research by IRB is required for US studies funded by the Department of Health and Human Services (DHHS) and other US federal agencies, as well as for research testing interventions—such as drugs, biologics, and devices—that are under the jurisdiction of the US Food and Drug Administration (FDA) (Table)
Regulation
Requirements
Membership (45CFR.46 107; 21CFR.56.107)
At least 5 members of varying backgrounds, both sexes, and > 1 profession
At least 1 scientific member, 1 nonscientific member, and 1 unaffiliated member
Members sufficiently qualified through diverse experience and expertise to safeguard subjects’ rights and welfare and to evaluate research acceptability related to laws, regulations, institutional commitments, and professional standards
At least 1 member knowledgeable about any regularly researched vulnerable groups
Members report and recusal for conflicts of interest Ad hoc experts as needed
Functions/operations (45CFR.46 108; 21CFR.56.108)
Follow written procedures for initial and continuing review and for any changes and amendments
Written procedures for reporting unanticipated problems, risks, and noncompliance Quorum of majority at convened meetings. Approval requires majority vote
Review (45CFR.46 109; 21CFR.56.109)
Authority to approve, require modifications of, or disapprove research
Require informed consent and documentation (or approve a waiver1)
Notify investigators in writing
At least annual continuing review
Criteria for approval (45CFR.46 111; 21CFR.56.111)
IRB should determine that risks are minimized; risks are reasonable in relation to anticipated benefits, if any, and the importance of the expected knowledge; subject selection is equitable and attention to vulnerable populations; informed consent will be sought and documented; adequate provisions for monitoring; adequate provisions to protect confidentiality; additional safeguards for subjects vulnerable to coercion or undue influence
Authority (45CFR.46. 113; 21CFR.56.113)
Institutional officials cannot approve research that is disapproved by the IRB (45CFR.46 only) The IRB can suspend or terminate research for serious harm or noncompliance
Records (45CFR.46. 115, 21CFR.56.115)
Records of research proposals, meetings, actions, correspondence, members, and so forth
CFR = Code of Federal Regulations
Where Does an IRB Get Its Authority?
In 1974, the Department of Health Education and Welfare instituted the regulations that formed the IRB for the protection of human subjects. IRBs are managed at state level by the Office for the Protection of Human Studies, an agency of the Department of Health and Human Services(OHRP).
OHRP supports IRBs in their work and receives allegations of inappropriate research practices and investigates them.
The institution served by the IRB offers administrative support for its operations including the appointment of an person to supervise research and IRB functions within the institution.
The organization also files a “Assurance” with the federal government, explaining the protocols and instructions to be followed by the IRB.
What is an “assurance” or a “multiple project assurance?”
An “assurance,” is a contract signed in compliance with HHS regulations between an entity and Department of Health and Human Services (HHS).
Of work involving human subjects performed by HHS or funded in full or in part by HHS, the HHS regulations include the performance site institution’s written guarantee that the institution will comply with the HHS regulations on the security of human subjects[45 CFR part 46].
The assurance mechanism is described in 45 CFR 46.103. Once an institution’s assurance has been approved by the HHS, a number is assigned to the assurance.
Assurance can be for a single grant or contract (“single project assurance”); multiple grants (“multiple project assurances”-commonly known as “general assurances”); or for other forms of projects, such as oncology group studies and AIDS research group studies (“cooperative project assurances”).
The Office for Human Research Protection (OHRP) is responsible for implementing the HHS regulations.
Is an “assurance” required by FDA?
At present, FDA regulations do not require an assurance. FDA regulations [21 CFR parts 50 and 56] apply to the research involving products regulated by FDA – federal funds and/or support do not need to be involved for the FDA regulations to apply.
IRBs are responsible for reviewing all federally funded research projects involving human subjects with a few exceptions ( e.g., evaluations of documents or surveys in which subjects cannot be identified individually or where exposure of the responses of respondents does not put them at risk of criminal or civil liability and does not affect participants financially, professionally or socially).
On the other hand, based on the procedures set forth in the Institutional Assurance, the IRB may review all the research projects, regardless of the source of funding.
How Does an IRB Make Its Decisions?
Before an investigator can receive federal funds in order to conduct a research project, the protocol (research procedures) is reviewed by the IRB.
The researcher provides the IRB with all the necessary materials for conducting its review, including a full description of the proposed project, materials that will be used by the subjects (surveys, question, tests, etc.),), a description of how the subjects will be recruited and consent to participate in the project (including a consent form), and how confidentiality of the subjects will be maintained.
The IRB analyses all of these materials to determine if there is sufficient protection for the research participants. Consideration by the IRB is largely focused on assessing the research’s costs and benefits.
Risks may be physical , social, psychological or economic. Benefits include both those for the individual person in the study and for the whole of society.
The IRB also considers the population being studied — Does it require additional protections? Would this population assess the risks and the benefits differently?
What Does An IRB Do After Reviewing the Project?
After evaluating the materials that the researcher offers to the IRB, they have to decide if the research ‘s benefits have been maximized and the risks minimized, and make a final decision as to whether the benefits justify the risks to the subject.
If the IRB considers this to be the case, then the protocol can be accepted. Alternatively, the IRB may demand that the researcher make specific methodological changes and approve the protocol based on those changes, or recommend revision and re-submission of the protocol.
The IRB can ultimately decide to disapprove of the proposal. Institutional officials may reject IRB-approved research protocols but may not grant approval for research projects that have been disapproved by the IRB.
In addition to evaluating new research procedures, IRBs are also monitoring existing programs or those that have process changes. Continuing projects are reviewed yearly (or more often if the IRB feels it is necessary).
How Does an IRB Protect Special Populations?
The Code of Federal Regulations requires IRBs to give special consideration to several classes of subjects: children, prisoners, pregnant women, mentally disabled people and economically or educationally disadvantaged people.
There are a number of ways the IRB carries out this charge. In certain cases, work with such subjects will only be accepted by the IRB when it requires minimal risk or when the benefits relate directly to the subject.
Furthermore, if the IRB regularly examines procedures that include one of the special groups, they might have a community member whose main concerns are in one of those groups.
Who has Access to IRB Records?
The institution and the IRB maintain records of IRB practices including copies of the checked work procedures, minutes of meetings, and correspondence. All documents must be made accessible by OHRP for analysis.”
August 14, 2020: “AstraZeneca has concluded an agreement with the European Commission (EC) to supply up to 400 million doses of the AZD1222 COVID-19 vaccine.
Building on the existing agreement with Europe’s Inclusive Vaccines Alliance spearheaded by Germany, France, Italy and the Netherlands, this new agreement will give all EU member states the option to access the vaccine in an equitable manner at no profit during the pandemic.
It also allows EU member states to redirect doses to other European countries.
Pascal Soriot, Chief Executive Officer, said: “This first vaccine agreement with the European Commission will ensure that millions of Europeans have access to the AZD1222 vaccine following its approval.
With production in our European supply chain soon to be started, we hope to make the vaccine available widely and rapidly, with the first doses to be delivered by the end of 2020.
I would like to thank the entire European Commission, and especially the Commissioner for Health and Food Safety, Stella Kyriakides, for their swift response in ensuring Europeans may soon be protected with a vaccine against this deadly virus, enabling our global society and economy to rebuild.”
In July 2020, interim results from the ongoing Phase I/II COV001 trial were published in The Lancet and showed AZD1222 was tolerated and generated robust immune responses against the SARS-CoV-2 virus in all evaluated participants.
Clinical development of AZD1222 is progressing globally with late-stage Phase II/III trials ongoing in the UK and Brazil, a Phase I/II trial in South Africa, and trials planned in the US, Japan and Russia.
Results from the late-stage trials are anticipated later this year, depending on the rate of infection within the clinical trial communities.
AstraZeneca continues to engage with governments, multilateral organisations and partners around the world to ensure broad and equitable access to the vaccine, should clinical trials prove successful.
Recent supply announcements with Russia, South Korea, Japan, China, Latin America and Brazil take the global supply capacity towards three billion doses of the vaccine.
AZD1222
AZD1222 was co-invented by the University of Oxford and its spin-out company, Vaccitech.
It uses a replication-deficient chimpanzee viral vector based on a weakened version of a common cold virus (adenovirus) that causes infections in chimpanzees and contains the genetic material of the SARS-CoV-2 virus spike protein.
August 14, 2020: “The U.S. Food and Drug Administration announced the following actions taken in its ongoing response effort to the COVID-19 pandemic:
The FDA is providing a device shortage list as part of the implementation of section 506J of the Federal Food, Drug, and Cosmetic Act (FD&C Act). The device shortage list reflects the categories of devices that the FDA has determined to be in shortage at this time, and will be updated as the COVID-19 pandemic evolves.
In addition, the FDA is providing a list of medical devices for which manufacturing has been permanently discontinued. Under section 506J, manufacturers of certain devices must notify the FDA of an interruption or permanent discontinuance in manufacturing.
The publication of these lists allows for transparency to the public and stakeholders about devices shortages and manufacturing that has been permanently discontinued.
The FDA issued an updated FDA COVID-19 Response At-A-Glance Summary that provides a quick look at facts, figures, and highlights of the agency’s response efforts.
The FDA issued an Emergency Use Authorization (EUA) for the emergency use of Baxter Healthcare Corporation’s REGIOCIT for adult patients being treated with continuous renal replacement therapy (CRRT) and for whom regional citrate anticoagulation is appropriate.
The use of this product under the EUA is limited to critical care settings. CRRT is a “dialysis” treatment that provides renal support for critically ill patients with acute kidney injury.
Baxter Healthcare Corporation’s REGIOCIT is available for use only in healthcare facilities that the company has qualified for receiving this product.
Testing updates:
To date, the FDA has currently authorized 213 tests under EUAs; these include 174 molecular tests, 37 antibody tests, and 2 antigen tests.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices.
The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.”







 Update: Daily Roundup August 17, 2020")