
What you need to include in a research paper is ABSTRACT:
Now, why is the abstract important?: It essentially seduces the reader into reading the rest of your paper and provide a general understanding about the story inside. An abstract describes about what you do in your essay, whether it’s a scientific experiment or a literary analysis paper. Your abstract must summarize all of the parts of the paper by writing accurately, concisely, and includes only the most important content so that you can keep those readers seduced into reading your paper.
To write an abstract, conclude your paper first, then type a summary that defines the purpose, problem, methods, results, and conclusion of your work. After you get the details down, all that’s left is just to format it correctly. Since an abstract is only a summary of your workdone, so it’s easy to accomplish!
So, before coming to structure of the abstract let’s talk about the couple of things you need to keep in mind before you begin to write it.
Getting your abstract started:
1. Write your paper first: Although an abstract goes at the beginning of the work, it acts as a summary of your entire paper. Put away writing your abstract for last, after you have already finished your paper.
2. Review and try to understand any requirements for writing your abstract. The paper you are writing probably has some specific guidelines and requirements, whether it is for publication in a journal, submission in a class, or part of a work project. Before you start writing, first refer to the guidelines, also about the LENGTH,STYLE and REQUIRENMENTS.
3. Consider your audience. Refer to others persons as guide or mentor just to know, what changes you still have to do.
4. Verify the type of abstract.
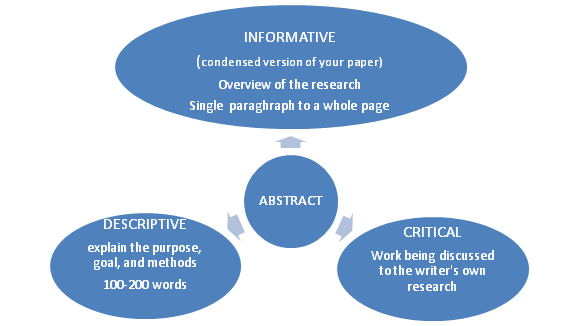
Now start writing your Abstract: There are total 5 parts of abstract, as given below. Purpose
Problem
Method
Result
conclusion
1. Identify your purpose. The reader wishes to know why your research is important, and what’s the purpose of it. Example: “This study explains about Development and Characterization of the Paclitaxel loaded Riboflavin and Thiamine Conjugated Poly (propylene Imine) Dendrimer mediated brain delivery”
2. Explain the problem. Abstracts state the “problem” behind your work. What problems is your research trying to better understand or solve, also what is the scope , What are your main claim or argument? “Brain tumor is difficult to treat because of poor absorption of many potentially useful antitumor drugs through BBB (blood brain barrier). Present study was aimed for developing and exploring the use of Paclitaxel- loaded Riboflavin and Thiamine Conjugated Poly (propylene Imine) Dendrimer for increase delivery of this drug across BBB.”
3. Explain your methods. Now this is the part where you give an overview of how you gifted your study. If you did your own work then include a description of it or If you reviewed the work of others, it can be explained in few words. “The developed conjugates were characterized using FTIR, NMR spectroscopy, electron microscopy drug loading, release, hemolytic, and ex vivo studies.”
4. Describe your results (informative abstract only). This only for informative abstract, you will be asked to present the results of your study. What you found(results)?, What answer did you attain from your research or study?, Was your theories or argument supported?, and what are the general findings? “The PTX-Tm-PPI and PTX-Rf-PPI showed more cytotoxic effect as compared to PTX and PTX-PPI with PTX-Rf-PPI exhibiting the maximum cytotoxic potential. Biodistribution studies confirmed about the targeting efficiency and higher biodistribution of Tm-PPI conjugates into the brain.”
5. Give your conclusion (in both descriptive and informative abstracts). This should end up your summary and give closing to your abstract. In it, deal with the meaning of your findings as well as the importance of your overall paper. “The result concluded that the riboflavin and thiamine conjugated PPI dendrimer has great potential to deliver significantly higher amount of drug to brain tumor for improved therapeutic outcome.”
Formatting Your Abstract:
1.Keep it in order. Write your abstract in an order like first write introduction, body, and then conclusion. Many journals have specific style guidelines for the abstracts. If you have been given a set of rules or guidelines, follow them to the letter.
2. Provide helpful information. Unlike a topic paragraph, which may be intentionally indefinite, an abstract should provide helpful explanation of your paper and your research. Try to avoid using direct acronyms or abbreviations in the abstract and do not include tables, figures, sources, or long quotations in your abstract. “Brain tumor is difficult to treat because of poor absorption of many potentially useful antitumor drugs through BBB (blood brain barrier). Present study was aimed for developing and exploring the use of Paclitaxel- loaded Riboflavin and Thiamine Conjugated Poly (propylene Imine) Dendrimer for increase delivery of this drug across BBB.”
3. Write it from scratch. Although your abstract is a summary but it should be written entirely separate from your paper. Don’t just copy-paste original sentences from paper, also avoid simply paraphrasing your own sentences, try to write your sentences using completely new vocabulary and phrases just to keep it interesting.
4. Use key words and phrases. If you want your abstract is to be published in a journal and people should be able to find it easily, then readers will search for definite queries on online databases in hopes that the papers, like yours, will show up. Also, try to use 5-10 important words or phrases key to your research in your abstract.
KEY WORDS PPI Dendrimer. paclitaxel. riboflavin. thiamine. Vitamins
5. Use real information. You want to represent people in with your abstract; it is the hook and eye that will promote them to continue reading your paper. On the other hand, do not reference ideas or studies that you don’t include in your paper to do this. Citing material that you don’t use in your work will misinform readers and ultimately lower your viewership.
6. Avoid being too specific. An abstract is a summary , you should not need to explain or define any terms in your abstract, a reference is all that is needed. Avoid being too explicit in your summary and stick to a very broad overview of your work and avoid terminologies. https://www.ncbi.nlm.nih.gov/pubmed/22994427
https://www.wikihow.com/Write-an-Abstract


 in combination with erbitux® (cetuximab) (braftovi doublet) for the treatment of Brafv600e-mutant metastatic colorectal cancer after previous therapies")


 for CAR T-Cell Therapy Lisocabtagene Maraleucel (liso-cel)")
 for the treatment of locally Advanced or Metastatic Urothelial Cancer")
 LUSTER Phase III studies in patients with uncontrolled GINA 4/5 asthma")

 Treatment")