
50 regulatory authorities from the 31 European Economic Area (EEA) countries (28 EU Member States, including Iceland, Liechtenstein and Norway), the European Commission and EMA creates the European medicines regulatory system.
European medicines regulatory system= (European commission + EMA + 50 Regulatory authorities)
This network makes EU regulatory system unique and quite complex. Although this is not its official name, the EMA is also called the European Medicines Assessment Agency or EMEA. It is currently called The European Agency for Medicines.
Table of Contents
Before Proceeding into approval process, let’s learn about two common terminologies.
Clinical Trial Application (CTA). It consists of comprehensive information about the investigational medicinal product and details about the proposed/planned trial. Regulatory authority reviews this application and decides about acceptability of conducting a trial.
It can be considered as equivalent to IND (Investigational new drug application) which needs to be submitted to FDA to get permission to initial a trial.
Marketing Authorization Application (MAA): It is an application submitted by a drug manufacturer/sponsor seeking permission to bring a drug product to the market. It can be consider as Equivalent to NDA of US regulatory
The below information covers only drug for human use.
There are four approval paths:
- Centralised procedure
- Decentralised procedure
- Mutual-recognition procedure
- Nationalized Procedure
Centralized procedure:
- It allows the marketing of a drug candidate on the basis of a single EU-wide assessment and marketing authorisation which is valid throughout the EU (European Union).
- Pharmaceutical companies or Sponsor has to submit single application to EMA and The Agency’s Committee for Medicinal Products for Human Use (CHMP) carries out a scientific assessment of the application and gives a recommendation to the European Commission. Based on the recommendation, marketing authorisation can be granted or rejected.
- Once it is approved, it is valid for all EU member state. The whole process may take around 210 days
Important Note:
The centralized procedure is compulsory in below scenario:
If Human Medicine contains new active substance to treat
- Cancer, Diabetes, Auto-immune disease, Orphan medicines, products manufactured by mean of biotechnology processes, neurodegenerative disorder, other immune dysfunctions, HIV and viral diseases.
- Gene-therapy, somatic cell-therapy or tissue-engineered medicines;
It is optional for other medicines:
- New active substances for indication other than listed above
- Significant therapeutic, scientific or technical innovation;
- Whose authorisation would be in the interest of public health at EU-Level.
Decentralised procedure:
- In this procedure the applicants/sponsor can apply for marketing authorization in more than one European Union Member States for those products which have not been yet authorized in any European Union country and also do not come under the list of drugs under compulsory Centralized Procedure category (List mentioned above under “important note”)
- The whole process may take around 210 days
Mutual Recognition Procedure:
Where applicant/Sponsor that have a drug authorized in one EU member states, can apply for this authorization to be recognized in other EU countries. Member States has to rely on each other’s scientific assessments
- The member state, where drug is already approved, is called reference member state and concerned member states are the one where applicant is seeking the approval through this process.
- This whole process can take up to a time period of 390 days.
Nationalized Procedure: In this procedure the applicant is allowed to obtain a marketing authorization in any of the one member states. To get the National Marketing authorization, the applicant needs to submit the application to the competent authority of the member state. It may take up to 210 days.
Generic and Hybrid Medicine approval process:
Generic Medicine: As per EMA:
“A generic medicine contains the same active substance(s) as the reference medicine, and is used for treating the same disease(s) at the same dose(s). Nevertheless, the inactive ingredients of a generic drug can be specific in name, appearance and packaging.”
Since the safety and efficacy data of the active substance is already available from the reference drug, sponsor/applicant only need to product below information:
- information on the quality of the medicine;
- bioequivalence study
Conceptually It is like FDA’ 505 (j) pathway for Generic Drug approval with no Changes or few allowed changes.
Hybrid medicines: As per EMA:
- Authorization of Hybrid medicines depends partly on the results of tests on the reference medicine and partly on new data from clinical trials (New + Old Data)
Hybrid Medicines carries the same active ingredient (as Reference Medicine) but it but has a different strength/route of administration or a slightly different indication from the reference medicine.
- Additional Clinical trial may be required to test efficacy and/or safety.
- Conceptually It is like FDA’ 505 b(2) pathway for generic drug with Some changes.
List of Scientific Committees which do the scientific assessment.

- The member state, where the clinical trial is taken place, will be responsible for authorisation and oversight of a clinical trial.
- The European Clinical Trials Database (EudraCT) tracks all the authorized clinical trial in EU.
Ref: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/generic-hybrid-medicines
https://www.ema.europa.eu/en/documents/leaflet/european-regulatory-system-medicines-european-medicines-agency-consistent-approach-medicines_en.pdf

 approved sNDA to Revise Flexion Therapeutic’s ZILRETTA® (triamcinolone acetonide extended-release injectable suspension) for the treatment of osteoarthritis (OA) knee pain")

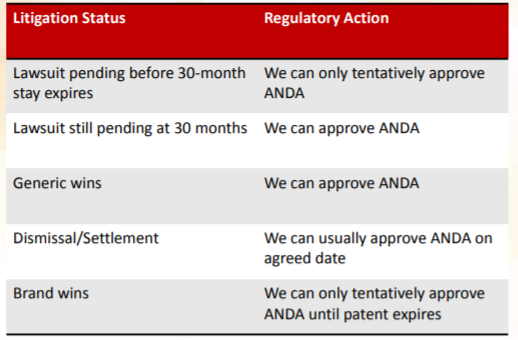
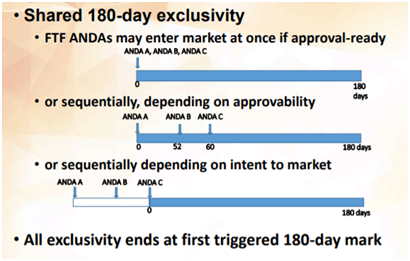
: Paragraph Certification I, II, III, and IV:")
 approved Allergan’s UBRELVY(ubrogepant) for the acute treatment of migraine with or without aura in adults.")
 for treatment of insomnia characterized by difficulties with sleep onset and/or sleep maintenance in adults")

")
