Jan 20, 2020:Heroic-Faith Medical Science expects its AI-powered respiratory monitor in order to receive US FDA clearance this year, according to Taiwan-based startup.
Heroic-Faith said the AI-powered nonstop respiratory monitor can perform the breathing sounds auscultation precisely, using Taiwan’s cutting-edge noise-cancelling technology, but also count breathing rate and identify the so-called adventitious sounds caused by the airway obstruction, spasm, and increased secretion, credit to the advanced AI algorithm.
Medical personnel can listen to the breathing sounds broadcasting from the device or through Bluetooth earphones in real-time, and meanwhile, visually monitor the sound spectrogram from the screen.
The alert system additional assists clinicians in early detection of respiratory compromises, according to the company.
The National Medical Products Administration (NMPA) ( Chinese name translation in English “State Drug Administration” or China Food and Drug Administration CFDA) and the Ethical Committee must approve a clinical trial application prior to the sponsor initiating a clinical trial.
First EC approves the application and NMPA’s approval depends on it. China Follows the centralized process for ethical review of the clinical trial application.
The ethical review process has three layers: national EC, provincial ECs, and institutional level ECs.
China’s National Health Commission (NHC) is responsible for managing ECs nationwide by establishing the National Committee of Medical Ethics Experts which develop policies for ethical review.
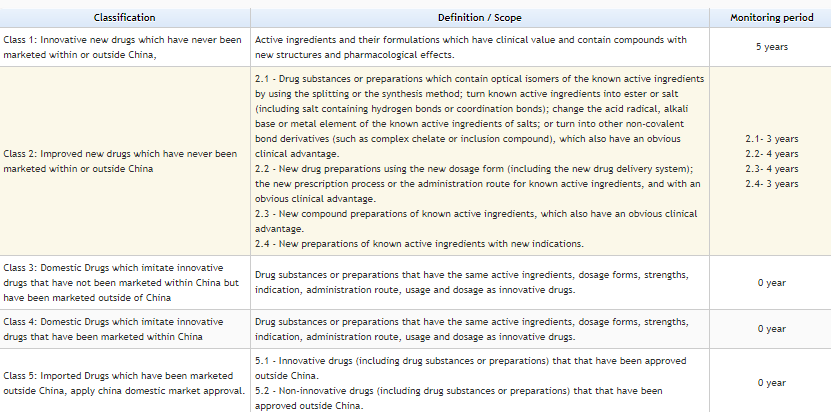
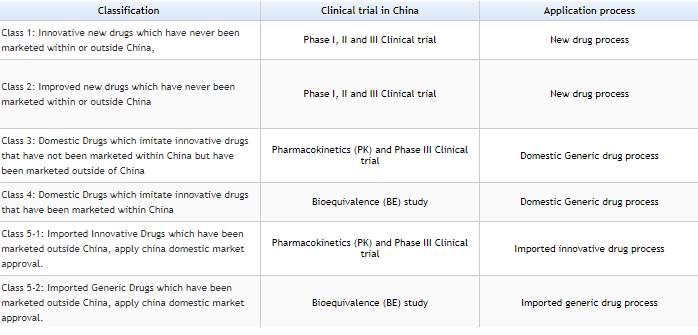
Please see the below tables for a comprehensive view of the drug approval process. The drugs are characterized into 5 Categorize as per New regulation by CFDA on Mar-2016. Credit: https://www.sfdachina.com/info/202-1.htm
In 1989, first CAR-T cells were developed by Gideon Gross and Zelig Eshhar at Weizmann Institute, Israel.
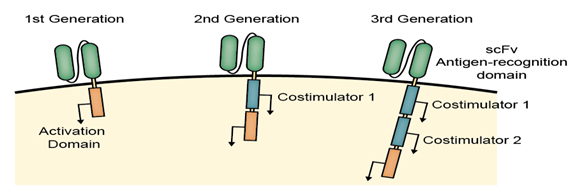
The complexity of the engineered CAR receptors has evolved over time, and depending on their composition, they are referred to as first, second , third, or fourth generation CARs.
Since the conventional cornerstones of cancer treatment (surgery, chemotherapy, and radiation therapy) manifest more and more limitations against multiple human malignancies, the novel immunotherapy is growing excitement with stutter steps, particularly the CAR-T therapy.
Hailstones as “a living drug”, CAR-T is an admirable example of the application of basic research to the clinic.
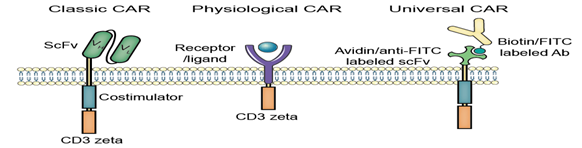
Representative CARs
The first fusion receptors
CAR stands for chimeric antigen receptor or artificial T cell receptor, which is engineered abundantly and endowing an immune effector cell (T cell) with an uninformed specificity via a monoclonal antibody.
The original CAR-T or the first-generation CAR consists of the intracellular domain from the CD3 ζ- chain and the primary transmitter of signals from the endogenous TCRs, which showed success in pre-clinical trials and entered in Phase I clinical trials in ovarian cancer, neuroblastoma and various types of leukaemia and lymphoma.
Even though the anti-tumor activity was limited due to insufficient activation, determination and homing to the cancer tissue, some significant effects certainly existed in patients with B-cell lymphoma treated with α-CD20-CD3 ζ CAR-modified T cells and also some neuroblastoma treated with ScFv-CD3 ζ CAR-Ts.
This is the most common form of CARs in order to fuse single-chain variable fragments (scFv) derived from monoclonal antibodies to CD3 ζ transmembrane and endo domain.
To augment the antitumor efficiency of 1st-generation CARs, the 2nd-generation CARs were designed to combine the intracellular signaling domains from various co stimulatory protein receptors (e.g., CD28, 41BB, ICOS) that are incorporated in the cytoplasmic tail of the CAR to enhance the signaling.
For instance, the CD19-targeted CARs incorporated with CD28 or 4-1BB signaling domains manifested outstanding complete remission rates in patients with refractory B-cell malignancies.
consequently, the CD28-based CARs showed a brisk proliferative response and boost effector functions. Meanwhile, the 4-1BB-based CARs manifested a other progressive T cell accumulation.
The three generations of CARs
As the expectation of more antitumor efficiency, the 3rd-generation of CARs combined with multiple signaling domains (e.g., CD3 ζ-CD28-41BB, CD3 ζ-CD28-OX40) to acquire additional enhanced activation signals, proliferation, production of cytokines and effective function.
For instance, the α-CD19-CD3 ζ-4-1BB CAR-Ts for chronic lymphocyte leukemia illustrates complete remission to infiltrate and lyse cancer tissue. Even better, a portion of CAR-Ts functioned as a memory phenotype for preventing the tumor relapses.
Despite the significant therapeutic effect, the emerging unmanageable activity accompanied with more antitumor efficacy caused life-threatening lysis activity as the most critical adverse effect or toxicity including clinically significant release of pro-inflammatory cytokines, pulmonary toxicity, multi-organ failure, and ultimate death.
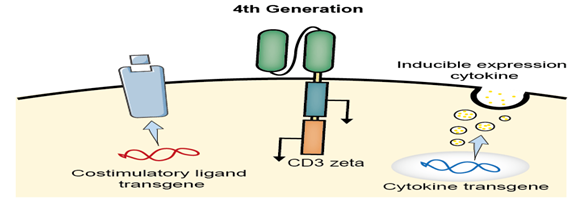
The 4th generation CAR
The previous CAR strategies are extremely specific and useful in redirecting T cells targeting malevolent cancer cells. However, the major limitation on solid tumors with a tremendous phenotypic heterogeneity and relapse due to antigen-negative cancer cells is the huge confront to trigger a novel CAR strategy.
The 4th-generation CAR-T is designed to shape the tumor environment by the inducible release of the transgenic immune modifiers, such as IL-12, which augments T-cell activation, attracts and activates native immune cells in order to eliminate antigen-negative cancer cells in the targeted lesion.
Control mechanisms: Adding a synthetic control mechanism to engineered T cells allows doctors to precisely control the persistence or activity of the T cells in the patient’s body, with the goal of reducing toxic side effects.
The major control techniques that trigger T cell death or limit T cell activation, and often regulate the T cells via a separate drug that can be introduced or withdrawn as needed.
Suicide genes: Heritably modified T cells are engineered in order to include one or more genes that can induce apoptosis when activated by an extracellular molecule.
Herpes simplex virus thymidine kinase (HSV-TK) and inducible caspase 9 (iCasp9) are two types of suicide genes that have been incorporated into CAR-T cells.
ON-switch: In this system, CAR-T cells can only function in the existence of both tumor antigen and a benign exogenous molecule. To achieve this, the CAR-T cell’s engineered chimeric antigen receptor is rip into two separate proteins that must come together to function.
The first receptor protein usually contains the extracellular antigen binding domain, while the second protein contains the downstream signaling elements and co-stimulatory molecules (such as CD3ζ and 4-1BB).
In the presence of an exogenous molecule (such as a rapamycin analog), the binding and signaling proteins dimerize collectively, allowing the CAR-T cells to attack the tumor.
Small molecule drug conjugates adaptor technology
SMDCs (small molecule drug conjugates) platform in immuno-oncology is an experimental approach that makes feasible the engineering of a single universal CAR T cell, which binds with extraordinarily high resemblance to a benign molecule designated as fluorescein isothiocyanate (FITC).
Also these cells are used to treat various cancer types when co-administered with bispecific SMDC adaptor molecules. These unique bispecific adaptors are constructed with a FITC molecule and a tumor-homing molecule to accurately bridge the universal CAR T cell with the cancer cells, which causes localized T cell activation.
Anti-tumor activity in mice is induced only when both the universal CAR T cells and the correct antigen-specific adaptor molecules are present. By adjusting the administered adaptor molecule dosing, anti-tumor activity and toxicity can be controlled.
Treatment of antigenically varied tumors can be achieved by administration of a mixture of the desired antigen-specific adaptors. There are several challenges of current CAR T cell therapies, such as:
the inability to control the rate of cytokine release and tumor lysis
the absence of an off switch that would terminate cytotoxic activity when tumor abolition is complete
Jan 16, 2020: WuXi Biologics and Bayer jointly announced an acquisition agreement that WuXi Biologics Germany GmbH will take over the operations of one of Bayer’s final drug product manufacturing plants in Leverkusen, Germany, and purchase the related equipment, in combination with a long-term lease contract for the building.
Based on the manufacturing agreement to be negotiated, the plant would be operated by the WuXi Biologics and serve as a back-up site for final product manufacturing of Kovaltry™, an antihemophilic factor (recombinant).
The transaction is expected to be concluded in the coming months subject to the satisfaction of the customary closing conditions. Financial details were not disclosed. https://www.wuxibiologics.com/wuxi-biologics-bayer-enter-acquisition-agreement-drug-product-plant-germany/
Jan. 16, 2020: TG Therapeutics, Inc. announced that the Company has been initiating a rolling submission of a New Drug Application (NDA) to the U.S. FDA requesting accelerated approval of umbralisib, oral, once-daily, dual inhibitor of PI3K-delta and CK1-epsilon, as a treatment for the patients with previously treated marginal zone lymphoma (MZL) and follicular lymphoma (FL).
The Company has received guidance from the FDA that the submission of a single NDA for both the MZL and FL indications is acceptable. Umbralisib has earlier been granted both orphan drug designation and breakthrough therapy designation by the FDA for MZL.
The Company expects to complete the NDA rolling submission with the first half of 2020.The UNITY- NHL trial is the multicenter, open-label Phase 2b trial.
The Marginal Zone Lymphoma (MZL) cohort was designed in order to evaluate the safety and efficacy of single agent umbralisib, in the patients with MZL who have received at least one prior anti-CD20 regimen.
The primary endpoint is overall response rate (ORR) as determined by the central Independent Review Committee (IRC) assessment.
The Follicular Lymphoma (FL) cohort was designed in order to evaluate the safety and efficacy of single agent umbralisib in the patients with FL who have received at least two prior lines of the therapy, including an anti-CD20 regimen and an alkylating agent.
Jan. 16, 2020: Arena Pharmaceuticals, Inc. announced that the U.S. FDA granted Fast Track designation for APD418, a β3-adrenergic receptor (AdrR) antagonist and cardiac myotrope, in development for the treatment of decompensated heart failure (DHF). Around 10 million DHF patient hospital visits expected in the US by 2025 and few viable treatment options.
APD418 has the prospective to make a significant impact for these patients.APD418 is a β3-adrenergic receptor (AdrR) antagonist and cardiac myotrope for the decompensated heart failure (DHF).
Jan 14, 2020: The U.S. FDA is alerting the public that results from a clinical trial assessing safety shows possible increased risk of cancer with the weight management medicine Belviq, Belviq XR (lorcaserin).
The cause of the cancer is uncertain, and cannot concluded that lorcaserin contributes to the cancer risk. Clinical trial results evaluation will communicate the final conclusions and recommendations when completed our review.
Health care professionals should believe if the benefits of taking lorcaserin are likely to exceed the potential risks when deciding whether to prescribe or continue patients on lorcaserin.
Lorcaserin is a prescription medicine approved by FDA in 2012 for use with a reduced-calorie diet and increased physical activity in order to help weight loss in adults who are stout or are overweight and have weight-related medical problems.
Since a long time, Impact factor (IF) has been used as a key indicator of importance of a journal in a particular field. The founder of Institute for Scientific Information, Eugene Garfield has introduced it.
Let’s define it:
“In any given year, the impact factor of a journal is the number of citations, received in that year, of articles published in that journal during the two preceding years, divided by the total number of “citable items” published in that journal during the two preceding years.”
Impact factor (IF) of any journal can be calculated only after completing the minimum of 3 years of publication. It means it cannot be calculated for new journal which has not completed the desired period.
Example: 2018 impact factor =A/B Where A is the number of times articles published in 2017 and 2016 were cited by indexed journals during 2018. B is the total number of citable items like articles and reviews published by that journal in 2017 and 2016. The 2018 Impact factors are actually published in 2019.
In this case, Impact factor can be calculated once all of the 2018 publications have been processed by the indexing agency.
Significance and limitation of Impact factor:
Most frequent use of impact factor is in academic evaluation (in Universities and research institutes). It gives a general idea for public about prestige of journals.
It is quite useful in clarifying the significance of absolute (or total) citation frequencies.
It eliminates one important bias which tends to favor large journals over small ones, or frequently issued journals over less frequently issued ones. It also reduces the bias of selecting older journals over newer ones.
Inclusion of review articles or letters can influence a journal’s impact and its ranking in journal lists. Review articles generally are cited more commonly than typical research article thus impact factor may get increased.
Limitation of completion of 3 years of publication.
Agency which calculate the Impact factors: Clarivate Analytics
This agency began to publish Journal Citation Reports (JCR) in 1975 as part of the SCI (Science Citation Index) and the Social Sciences Citation Index (SSCI).
“The JCR provides quantitative tools for ranking, evaluating, categorizing, and comparing journals. Some of the journals listed in the JCR are not citing journals, but are cited-only journals. This is significant when comparing journals by impact factor because the self-citations from a cited-only journal are not included in its impact factor calculation (impact factor without self-citation).”
It is important to note that all journals are Not tracked in the JCR database. It means those journal do not have impact factor (IF)
Young or new journals has to wait until they have a record of citations before even being considered for inclusion
We cannot be 100 percent sure about scientific worth of an individual article or research by just looking the impact factor.
Jan 15, 2020: Bristol-Myers Squibb Company announced the U.S. Food and Drug Administration (FDA) has accepted its supplemental Biologics License Application (sBLA) for Opdivo (nivolumab) with Yervoy (ipilimumab) for the first-line treatment of patients with the metastatic or recurrent non-small cell lung cancer (NSCLC) with no EGFR or ALK genomic tumor aberrations.
The FDA has granted the application Priority Review with the Prescription Drug User Fee Act (PDUFA) aim date of May 15, 2020.This application is based on data from Part 1 of the Phase 3 CheckMate -227 trial evaluating Opdivo plus Yervoy vs. chemotherapy in patients with previously untreated NSCLC, in which the double immunotherapy combination demonstrated significant improvement in overall survival vs. chemotherapy alone. The safety profile of the Opdivo plus Yervoy was consistent with previously reported studies and no new safety signals were observed.
Jan 12, 2020: Imago BioSciences, Inc. announced that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation for the development of bomedemstat (IMG-7289) for the treatment of essential thrombocythemia (ET), a bone marrow disease linked with high platelet counts and potentially catastrophic vascular complications.
Bomedemstat inhibits the enzyme LSD1 (lysine-specific demethylase 1), as a result preventing excess platelet and neutrophil production.
The FDA grants Fast Track designation in order to facilitate development and expedite the review of therapies with the potential to treat a severe condition where there is an unmet medical need.
A therapeutic that receives Fast Track designation can
benefit from the early and frequent communication with agency, in addition to a
rolling submission of the marketing application, with the objective of getting
important new therapies to patients more speedily.
Bomedemstat is a small molecule that was discovered by Imago BioSciences, inhibits lysine-specific demethylase 1 (LSD1 or KDM1A), an enzyme essential for the production and normal function of megakaryocytes and for self-renewal of the malignant hematopoietic stem or progenitor cells.
Megakaryocytes are the primary producer of the platelets and cytokines that drive essential thrombocythemia pathogenesis.
Jan 13, 2020: MorphoSys AG and Incyte Corporation announced that the companies have entered into a collaboration and license agreement to further develop and commercialise MorphoSys’ proprietary anti-CD19 antibody tafasitamab (MOR208) globally.
Tafasitamab is an Fc-engineered antibody against CD19 currently in clinical development for the treatment of the B cell malignancies.
Both will co-commercialise tafasitamab in the US, while Incyte has exclusive commercialisation rights outside of the US.In the US, MorphoSys and Incyte will co-commercialize tafasitamab, with the MorphoSys leading the commercialization strategy and booking all the revenues from sales of tafasitamab.
They will be collectively responsible for commercialization activities in the US and will share profits and losses on a 50:50 basis.
Outside the US, Incyte will have exclusive commercialization rights, and will lead the commercialisation strategy and book all revenues from sales of tafasitamab, paying MorphoSys royalties on ex-US net sales.
Additionally, the companies will share development costs associated with global and US-specific trials at a rate of 55% (Incyte) to 45% (MorphoSys); Incyte will cover over 100% of the potential development costs for trials that are specific to ex-U.S. countries.
Both parties have agreed to co-develop tafasitamab broadly in relapsed/refractory diffuse large B cell lymphoma (r/r DLBCL), frontline DLBCL as well as additional indications beyond the DLBCL, such as the follicular lymphoma (FL), marginal zone lymphoma (MZL) and chronic lymphocytic leukemia (CLL).
MorphoSys recently submitted a Biologics License Application (BLA) for tafasitamab, in combination with lenalidomide, to the US Food and Drug Administration (FDA) for the treatment of r/r DLBCL; the FDA decision regarding a probable approval is expected by mid-2020.
Jan. 13, 2020: CytoDyn Inc. announced that the Company has filed for Breakthrough Therapy designation (BTD) with the U.S. Food and Drug Administration (FDA) for the use of leronlimab as adjuvant therapy for the treatment of metastatic triple-negative breast cancer (mTNBC).
The BTD filing is based on data as of the first patient in the Company’s mTNBC Phase 1b/2 trial and an additional single-patient trial under an emergency investigational new drug (IND) protocol evaluating leronlimab for the treatment of HER2+ metastatic, stage 4, breast cancer (MBC).
Data from the first patient in the Phase 1b/2 trial showed that the patient had no detectable circulating tumor cells (CTCs) or putative metastatic tumor cells in the peripheral blood and additional large reductions in CCR5 expression on cancer-associated cells at 11 weeks of treatment with the leronlimab.
This patient’s data also demonstrated tumor shrinkage of >20% after just a few weeks of treatment. The data from the patient under the emergency IND protocol with HER2+ metastatic, stage 4, MBC showed no sign of new metastatic spots in the liver, lung and brain during the treatment with leronlimab.
Breakthrough Therapy designation is a process designed to accelerate the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may display substantial improvement over available therapy on a clinically significant endpoint(s).
Triple-negative breast cancer (TNBC) is a type of breast cancer characterized by the absence of the three most common types of receptors in the cancer tumor known to fuel most breast cancer growth are estrogen receptors (ER), progesterone receptors (PR) and the hormone epidermal growth factor receptor 2 (HER-2) gene.






")



