
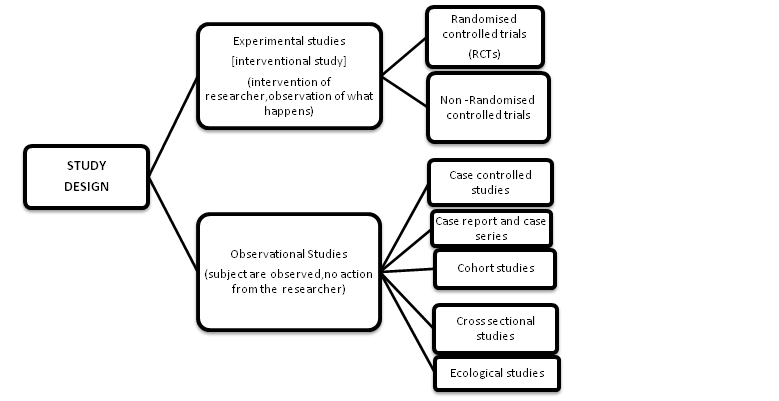
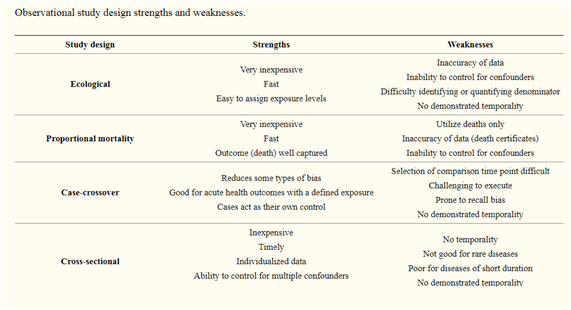
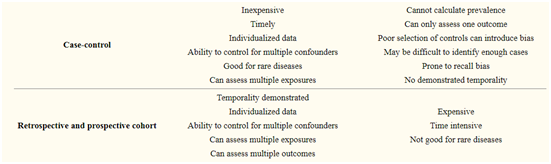
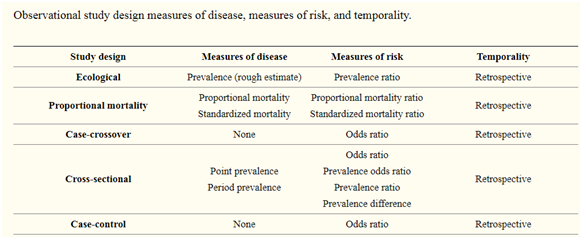
Table of Contents
May 11, 2020: “AstraZeneca and Daiichi Sankyo Company, Limited (Daiichi Sankyo)’s Enhertu (trastuzumab deruxtecan) has been granted Breakthrough Therapy Designation (BTD) in the US for the treatment of patients with HER2-positive unresectable or metastatic gastric or gastroesophageal junction adenocarcinoma who have received two or more prior regimens including trastuzumab.
Gastric cancer is the third leading cause of cancer mortality with a five-year survival rate of 5% for metastatic disease.
Approximately one in five gastric cancers are considered HER2 positive.
The Food and Drug Administration’s (FDA) BTD is designed to accelerate the development and regulatory review of potential new medicines that are intended to treat a serious condition and address a significant unmet medical need.
The new medicine needs to have shown encouraging early clinical results that demonstrate substantial improvement on a clinically significant endpoint over available medicines.
José Baselga, Executive Vice President, R&D Oncology, said: “Current therapy options are limited for patients with HER2-positive metastatic gastric cancer and for those who relapse, there are no approved HER2-targeted medicines.
We look forward to working with the FDA to further explore the potential of Enhertu to become an important new treatment and the first antibody-drug conjugate for this devastating disease.”
Gilles Gallant, Senior Vice President, Global Head, Oncology Development, Oncology R&D, Daiichi Sankyo, said: “DESTINY-Gastric01 represents the first randomised trial of Enhertu to demonstrate clinically meaningful and statistically significant results, including objective response and survival increases compared to physician’s choice of chemotherapy.
We are thrilled that the FDA has granted Enhertu a second Breakthrough Therapy Designation.”
The FDA granted BTD based on data from the registrational Phase II DESTINY-Gastric01 trial and data from the Phase I trial published in The Lancet Oncology.
In DESTINY-Gastric01, patients treated with Enhertu, a HER2-directed antibody drug conjugate (ADC), demonstrated a statistically significant and clinically meaningful improvement in objective response rate (ORR), the primary endpoint, and overall survival (OS), a key secondary endpoint, versus patients treated with investigator’s choice of chemotherapy (irinotecan or paclitaxel monotherapy).
The overall safety and tolerability profile of Enhertu (6.4 mg/kg) in DESTINY-Gastric01 were consistent with that seen in the Phase I trial.
The most common adverse events were hematologic and gastrointestinal including neutrophil count decrease, anaemia, nausea and decreased appetite.
There were cases of drug-related interstitial lung disease (ILD) and pneumonitis, the majority of which were Grade 1 and 2, with two Grade 3 and one Grade 4. No ILD-related deaths (Grade 5) occurred in patients with gastric cancer in the Phase I trial or in Phase II DESTINY-Gastric01 trial.
The full results of DESTINY-Gastric-01 will be presented during the 2020 American Society of Clinical Oncology ASCO20 Virtual Scientific Program.
Enhertu received SAKIGAKE designation in March 2018 by Japan’s Ministry of Health, Labour and Welfare (MHLW) for potential use in the same HER2-positive gastric cancer patient population and was recently submitted to the MHLW for approval.
This is the second BTD granted for Enhertu in the US. Enhertu received BTD in 2017 for HER2-positive metastatic breast cancer and received approval in December 2019.
Gastric (stomach) cancer is the fifth most common cancer worldwide and the third leading cause of cancer mortality; there were approximately one million new cases reported in 2018 and 783,000 deaths.
In the US, it is estimated that 27,600 new cases of gastric cancer will be diagnosed in 2020 and more than 11,000 people will die from the disease.
Approximately one in five gastric cancers are HER2 positive.
HER2 is a tyrosine kinase receptor growth-promoting protein expressed on the surface of many types of tumours including gastric, breast and lung cancers.
Gastric cancer is usually diagnosed in the advanced stage, but even when diagnosed in earlier stages of the disease the survival rate remains modest.
Recommended 1st-line treatment for HER2-positive advanced or metastatic gastric cancer is combination chemotherapy plus trastuzumab, an anti-HER2 medicine, which has been shown to improve survival outcomes when added to chemotherapy.
For gastric cancer that progresses on 1st-line treatment, there are no other approved HER2-targeting medicines.
DESTINY-Gastric01 is a registrational Phase II, open-label, multi-centre trial testing the safety and efficacy of Enhertu in a primary cohort of 188 patients from Japan and South Korea with HER2-expressing, advanced gastric cancer or gastroesophageal junction adenocarcinoma (defined as IHC3+ or IHC2+/ISH+) who have progressed on two or more prior treatment regimens including fluoropyrimidine and platinum chemotherapy and trastuzumab.
Patients were randomised 2:1 to receive Enhertu or investigator’s choice of chemotherapy (paclitaxel or irinotecan monotherapy).
Patients were treated with Enhertu 6.4 mg/kg once every three weeks or chemotherapy. The primary endpoint of the trial is ORR, as assessed by an independent review committee.
Secondary endpoints include OS, progression-free survival, duration of response, disease control rate and time to treatment failure as well as pharmacokinetic and safety endpoints.
Enhertu (trastuzumab deruxtecan; fam-trastuzumab deruxtecan-nxki in the US) is a HER2-directed ADC and is the lead ADC in the oncology portfolio of Daiichi Sankyo and the most advanced programme in AstraZeneca’s ADC scientific platform.
ADCs are targeted cancer medicines that deliver cytotoxic chemotherapy (“payload”) to cancer cells via a linker attached to a monoclonal antibody that binds to a specific target expressed on cancer cells.
Enhertu is approved in the US and Japan for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens based on the DESTINY-Breast01 trial.
A comprehensive development programme is underway globally with six registrational trials evaluating the efficacy and safety of Enhertu monotherapy across multiple HER2-driven cancers including breast, gastric and lung cancers.
Trials in combination with other anticancer treatments, such as immunotherapy, are also underway.
Collaboration between AstraZeneca and Daiichi Sankyo
In March 2019, AstraZeneca and Daiichi Sankyo entered into a global collaboration to jointly develop and commercialise Enhertu worldwide, except in Japan where Daiichi Sankyo maintains exclusive rights. Daiichi Sankyo is solely responsible for manufacturing and supply.“


 Update: FDA Authorizes First Antigen Test to Help in the Rapid Detection of the Virus that Causes COVID-19 in Patients")





 Phase 3 ADVANCE Study")